Clinical Description
The manifestations of malignant hyperthermia (MH) result from exposure to certain volatile anesthetic agents (i.e., halothane, isoflurane, sevoflurane, desflurane, and enflurane) that act as triggers either alone or in conjunction with succinylcholine, a depolarizing muscle relaxant. MH is an inherited pharmacogenetic disorder of calcium regulation resulting in uncontrolled skeletal muscle hypermetabolism [Rosenberg et al 2015] with variable clinical presentations (depending on the triggering agents and environmental factors, such as metabolic state and body temperature) at the beginning of anesthesia.
The triggering substances initiate uncontrolled release of calcium from the sarcoplasmic reticulum via the skeletal muscle calcium release channel (RyR1), and also may promote entry of extracellular calcium into the myoplasm leading to the sustained pathologic increase in cytosolic calcium in skeletal muscle cells [Yang et al 2007, Duke et al 2010, Riazi et al 2018]. Increased myoplasmic calcium causes contracture of skeletal muscles and activates glycogenolysis and cell metabolism, resulting in excessive production of heat and excess lactate. Activation of the oxidative cycle leads to high oxygen consumption and high carbon dioxide production.
MH clinical manifestations are variable; with prompt and rapid clinical response, some signs may not appear. Hypercapnia is common, as is tachycardia. Hyperthermia may be one of the early signs of MH. However, failure to monitor core temperature may lead to a delay in detecting hyperthermia. Skin temperature measurement is often misleading during MH crises [Larach et al 2010]. Acidosis may be mild if the syndrome is recognized and treated promptly. HyperCKemia and rhabdomyolysis are more common when succinylcholine has been used but may be mild or not appear at all in some individuals, for reasons that are not clear. In some instances rhabdomyolysis does not appear for several hours. Hyperkalemia, leading to cardiac arrhythmia and even arrest, is uncommon if the syndrome is detected and treated promptly but may develop with remarkable rapidity.
In survivors, normalization of edematous muscle and serum CK concentration occurs within ten to 15 days, but symptom resolution may take longer () [Jurkat-Rott et al 2000].
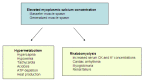
Clinical features of malignant hyperthermia susceptibility Note: Early diagnosis and rapid therapy are both life saving and lead to a reduction of clinical symptoms.
MH may appear at any point during anesthetization or within an hour or so after termination of anesthesia. If succinylcholine is used during induction of anesthesia, an acceleration of the manifestations of MH may occur; tachycardia, elevation of end-tidal carbon dioxide levels, hypertension, marked temperature elevation, and arrhythmias are seen over the course of five to ten minutes. However, a completely normal response to succinylcholine may be present in some individuals susceptible to MH; in these individuals, a potent inhalation agent is apparently necessary to trigger the syndrome.
In almost all instances, the first manifestations of MH occur in the operating room. In classic MH, the initial signs are tachycardia, rapidly rising end-tidal C02, and tachypnea. Tachypnea is usually not recognized because most individuals receiving general anesthesia are paralyzed. Shortly after the heart rate increases, the blood pressure may increase, often associated with ventricular arrhythmias induced by sympathetic nervous system stimulation from hypercarbia, hyperkalemia, and catecholamine release. Thereafter, muscle rigidity or increased muscle tone may become apparent; and body temperature increases at a rate of 1°-2° C every five minutes.
At the same time, the CO2 absorbent used in general anesthesia becomes activated and warm to the touch from the exothermic reaction with the CO2 exhaled by the affected individual. The individual may display peripheral mottling, on occasion sweating, and in rare cases cyanosis. Blood gas analysis usually reveals hypercarbia (PCO2>60 mm Hg) and respiratory and metabolic acidosis without oxygen desaturation. Elevation of end-tidal CO2 greater than 55 mm Hg is one of the earliest signs of MH; however, vigorous mechanical hyperventilation may prevent hypercarbia and delay the diagnosis [Karan et al 1994]. A mixed venous blood sample shows even more evidence of CO2 retention and metabolic acidosis. Hyperkalemia, hypercalcemia, lactacidemia, and myoglobinuria are characteristic but not always present. Increase in serum CK concentration often exceeds 20,000 units/L in the first 12-24 hours.
Death results unless the individual is promptly treated (see Management). Even with treatment and survival, the individual is at risk for life-threatening myoglobinuric renal failure, disseminated intravascular coagulation (DIC), compartment syndrome, and recrudescence of the syndrome within the first 24-36 hours following the episode. A study of MH using a North American MH registry containing information about affected individuals reported between 1987 and 2006 showed that nonfatal complications occurred in 35% of these individuals. Twelve of these complications included cardiac, renal, or hepatic dysfunction; coma or change in consciousness level; pulmonary edema; and DIC [Larach et al 2010].
Early diagnosis and rapid therapy are life saving and also lead to a reduction of clinical symptoms. It should be noted that modern anesthetic care and monitoring often allow early detection of MH. Treatment with dantrolene results in much lower morbidity and mortality than first reported when MH was recognized in the 1960s [Larach et al 2008]; however, mortality may be as high as 11% [Rosero et al 2009]. The likelihood of any complication increased 2.9 times per 2° C increase in maximum temperature and 1.6 times per 30-minute delay in dantrolene administration [Larach et al 2010]. The most frequent complications associated with dantrolene administration are muscle weakness (14.6%), phlebitis (9.2%), and gastrointestinal upset (4.3%). There is a 25% increase in the risk for any of the above complications when the total dose of dantrolene as required by clinical indications is twice the recommended initial treatment dose of 2.5 mg/kg [Brandom et al 2011].
The presentation of MH outside a hospital setting may pose special problems. Several deaths from MH have occurred when the episode began in an ambulatory surgery setting. Probable causes include inadequate preparation for treating MH (including absence of dantrolene), insufficient and unprepared personnel, and problems in stabilizing an affected individual prior to transfer to a hospital. It is suggested that all facilities have a plan to deal with MH and hold practice drills at regular intervals (see Larach et al [2012] for transfer-of-care protocols).
MH may also occur in the early postoperative period, usually within the first hour of recovery from anesthesia. Characteristic tachycardia, tachypnea, hypertension, and arrhythmias presage an episode of MH. Isolated myoglobinuria without an obvious increase in metabolism in the postoperative period (≤24 hours) should alert the anesthesiologist to the possibility of MH.
Of note, an MH episode may not occur with every exposure to "trigger" agents; clinical manifestations depend on genetic predisposition, dose of trigger agents, and duration of trigger exposure.
Signs of MH have also been reported without exposure to anesthetic agents. In some cases signs follow overdose of MDMA agonists; in other cases MH may be associated with heat and exercise.
Environmental/Exertional Heat Stress
Recent clinical, genetic, and laboratory studies using animal models provide evidence for a relationship between environmental or exertional heat stress (EHS) and MHS [Chelu et al 2006, Yang et al 2006, Durham et al 2008, Lanner et al 2012]. Some individuals who have experienced exertional heat illness have been found to be MH susceptible based on contracture testing [Capacchione & Muldoon 2009]. In one study, one third of young military recruits who experienced exercise-induced heat illness had an abnormal contracture response.
Evidence of a relation between EHS and MHS is presented by Tobin et al [2001] in the case report of a boy age 12 years who died from an MH-like event following participation in a football game. The boy had recovered from a previous clinical MH episode during general anesthesia with sevoflurane; sequence analysis revealed that both the boy and his father had a common RYR1 pathogenic variant (p.Arg163Cys). A more recent study found that two unrelated children who experienced fatal non-anesthetic awake episodes triggered by either a viral prodrome or exposure to environmental heat stress possessed an identical RYR1 variant (p.Arg3983Cys), while one of the children also had a second variant (p.Asp4505His) [Groom et al 2011].
MHS Phenotypes
Several distinct RYR1-related myopathies can predispose to classic MH:
Central core disease (OMIM
117000) and
multiminicore disease (OMIM
255320) are myopathies caused by mutation of
RYR1. Muscle weakness can range from mild to severe. Most affected individuals have mild disease with symmetric proximal muscle weakness & variable involvement of facial & neck muscles. Motor development is usually delayed, but most affected individuals acquire independent ambulation. Severe disease is early in onset with profound hypotonia often accompanied by poor fetal movement, spinal deformities, hip dislocation, joint contractures, poor suck, and respiratory insufficiency requiring assisted ventilation. Multiminocore disease is broadly classified into four groups: classic form, moderate form with hand involvement, antenatal form with arthrogryposis multiplex congenita, and ophthalmoplegic form. About 75% of affected individuals have classic symptoms characterized by neonatal hypotonia, delayed motor development, and axial muscle weakness, which leads to development of scoliosis and significant respiratory involvement; varying severity of spinal rigidity is present. Each of the other three forms is seen in fewer than 10% of individuals.
King or King-Denborough syndrome (OMIM
145600) is characterized by: distinctive facies, ptosis, downslanted palpebral fissures, widely spaced eyes, epicanthal folds, low-set ears, malar hypoplasia, micrognathia, high-arched palate, clinodactyly, single palmar crease,
pectus excavatum, winging of the scapulae, lumbar lordosis, and mild thoracic scoliosis. Individuals present with hypotonia at birth, slightly delayed motor development, diffuse joint hyperextensibility, and mild proximal muscle weakness. Muscle biopsy reveals minimal but identifiable changes represented by fiber size variability, type I fiber predominance and atrophy, perimysial fibrous infiltration, and some disarray of the intermyofibrillary network. Pathogenic variants in
RYR1 have been found in some individuals with King-Denborough syndrome.
STAC3 disorder
(Native American myopathy), caused by biallelic pathogenic variants in
STAC3, is characterized by congenital myopathy and musculoskeletal involvement of the trunk and extremities. Most children have weakness with myopathic facies, progressive kyphoscoliosis, and contractures. Other common findings are palatal anomalies (including cleft palate) and short stature. Risks for MHS and restrictive lung disease are increased. Intellect is typically normal.
Other RYR1 allelic conditions associated with MH susceptibility:
Vladutiu et al [2011] revealed that variants in
RYR1 may contribute to the underlying genetic risk for non-anesthesia-induced myopathies, such as statin-induced myopathy.
In a study of 12 young men with exercise-induced rhabdomyolysis (ER), ten were determined to be MH susceptible on contracture testing and three had known MHS
RYR1 pathogenic variants [
Wappler et al 2001]. In addition, the two
RYR1 pathogenic variants p.Arg401Cys and p.Arg614Cys are associated with MHS, EHS, and ER [
Davis et al 2002].
RYR1 variants have also been found to underlie ER in African American men [
Sambuughin et al 2009]. This study identified three novel
RYR1 variants: p.Ala933Thr, p.Gly2160Ser, and p.Thr4294Met, in individuals with ER.
Retrospective data on Canadian individuals with MHS and ER showed that an
RYR1 or
CACNA1S pathogenic variant was identified in three of 17 individuals [
Kraeva et al 2017].