Clinical Description
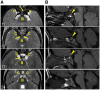
To date, 25 individuals have been identified with a germline (de novo or inherited from a mosaic parent) pathogenic C-terminal variant in MN1 [Mak et al 2020, Miyake et al 2020]. Individuals with MN1 C-terminal truncation (MCTT) syndrome have intellectual disability, severe expressive language delay, dysmorphic facial features (see ), and distinctive findings on brain imaging (including perisylvian polymicrogyria and atypical rhombencephalosynapsis) (see Table 1). The summary of the phenotypic features associated with MCTT syndrome in Table 3 is based on these reports.
Table 3.
Clinical Features of MN1 C-Terminal Truncation Syndrome
View in own window
| Feature | Persons w/Feature |
|---|
| Intellectual disability | 19/20 (95%) |
| Motor delay | 19/20 (95%) |
| Speech delay | 21/23 (91%) |
| Hypotonia | 19/21 (90%) |
| Hearing loss | 16/20 (80%) |
| Feeding difficulties | 14/21 (67%) |
| Spinal anomalies | 8/12 (67%) |
| Dental anomalies | 9/15 (60%) |
| Ophthalmologic anomalies | 10/17 (59%) |
| Cardiovascular anomalies | 6/20 (30%) |
| Seizures | 6/22 (27%) |
| Craniosynostosis | 3/22 (14%) |
Developmental delay (DD) / intellectual disability (ID). Individuals with MCTT syndrome have mild-to-moderate intellectual disability and severe expressive language delay. The majority have nonverbal communication with notable exceptions. For example, one child was able to speak at age two years, and at age 14 years was able to function at the level of a seven-year-old. For others, single-word speech began between ages three and six years. Some can communicate in sign language with up to 50 signs [Mak et al 2020].
Delay in gross motor development included hypotonia; at least four of 22 children walked independently by age two to three years. Others required orthotics or a wheelchair for mobilization. As for fine motor and self-help skills, most individuals require help with writing, feeding, or dressing [Mak et al 2020].
Hearing loss, when present, is mild to moderate and prelingual. It is usually bilateral, conductive, and/or sensorineural. Dysplasia of the cochlea, semicircular canals, and bony structures of the middle ear (e.g., incus) has been reported.
Feeding difficulties are more prominent early in infancy and may resolve after the first year of life. Hyperphagia has also been reported in three children [Miyake et al 2020].
Spinal anomalies include lordosis, scoliosis, or kyphosis, which may be detected clinically or by imaging.
Dental anomalies can include conical teeth, crowded teeth, and serrated teeth. Malocclusion has also been reported.
Ophthalmologic anomalies can include oculomotor defects, strabismus, and/or shallow orbits, giving the appearance of exorbitism.
Cardiovascular anomalies include atrial septal defect or ventricular septal defects.
Seizures are generalized and may be myoclonic in nature. Among the six individuals with seizures, the majority were isolated events and were responsive to anti-seizure medication. While polymicrogyria may be associated with increased risk of seizures, more information is needed.
Craniosynostosis. Individuals with MCTT syndrome are at increased risk for craniosynostosis with no specific pattern to the sutures affected. Although head circumference is normal in individuals with craniosynostosis, head shape is consistently affected. Skull shape anomalies that may be observed with or without underlying craniosynostosis include dolichocephaly, turricephaly, and/or bitemporal narrowing, plagiocephaly, and macrocephaly. Individuals with craniosynostosis are at increased risk for elevated intracranial pressure.
Behavioral problems. While some affected individuals may experience frustration due to poor verbal communication, no consistent behavior problems have been reported. The natural history into adulthood has not yet been delineated.
Growth. Growth parameters tend to remain within the normal range.
Prognosis. It is unknown if life span is affected in MCTT syndrome. One individual is alive at age 21 years [Mak et al 2020]. An unreported male is well at age 39 years [Angela Lin, personal commmunication], demonstrating that survival into adulthood is possible. Since many adults with disabilities have not undergone advanced genetic testing, it is likely that adults with this condition are unrecognized and underreported.