Summary
Clinical characteristics.
The phenotypic spectrum of mucopolysaccharidosis IVA (MPS IVA) is a continuum that ranges from a severe and rapidly progressive early-onset form to a slowly progressive later-onset form. Children with MPS IVA typically have no distinctive clinical findings at birth. The severe form is usually apparent between ages one and three years, often first manifesting as kyphoscoliosis, genu valgum (knock-knee), and pectus carinatum; the slowly progressive form may not become evident until late childhood or adolescence, often first manifesting as hip problems (pain, stiffness, and Legg Perthes disease). Progressive bone and joint involvement leads to short stature, and eventually to disabling pain and arthritis. Involvement of other organ systems can lead to significant morbidity, including respiratory compromise, obstructive sleep apnea, valvular heart disease, hearing impairment, visual impairment from corneal clouding, dental abnormalities, and hepatomegaly. Compression of the spinal cord is a common complication that results in neurologic impairment. Children with MPS IVA have normal intellectual abilities at the outset of the disease.
Diagnosis/testing.
The diagnosis of MPS IVA is established in a proband by identification of low N-acetylgalactosamine 6-sulfatase (GALNS) enzyme activity in cultured fibroblasts or leukocytes or by identification of biallelic pathogenic variants in GALNS on molecular genetic testing.
Management.
Treatment of manifestations: Enzyme replacement therapy (elosulfase alfa) is available, although the data on long-term effects of this treatment on the skeletal and non-skeletal features of MPS IVA are limited. Evaluation and management of individuals with MPS IVA are best undertaken by multiple specialists, coordinated by a physician specializing in the care of persons with complex medical problems. Physiatrists, physical therapists, and occupational therapists help optimize mobility and autonomy. Psychological support can optimize coping skills and quality of life; educational professionals can optimize the learning environment for a medically fragile individual. Upper-extremity management may include stabilizing external wrist splints or partial or complete wrist fusion. Surgical intervention is often required for lower-extremity malalignment, hip subluxation and/or hip pain, upper cervical spine instability, and/or progressive thoracolumbar kyphosis. Cardiac valve involvement may require valve replacement. Bacterial endocarditis prophylaxis is recommended for those with a prosthetic cardiac valve, prosthetic material used for cardiac valve repair, or previous infective endocarditis. Upper-airway obstruction and obstructive sleep apnea are managed by removal of enlarged tonsils and adenoids; diffuse narrowing of the airway may require positive airway pressure and/or tracheostomy. All affected individuals should receive influenza and pneumococcal immunizations as well as routine immunizations. Anesthesia management by an experienced anesthesiologist. Potential pre- and postoperative anesthetic concerns secondary to spine anomalies and difficult airway management need to be anticipated. Optimize nutrition and provide adequate vitamin D and calcium. The outcome following keratoplasty for corneal opacification varies. Dental care to prevent cavities and orthodontic management as needed. Hearing loss is often treated initially with ventilation tubes and later with hearing aids. School accommodations as needed to prevent physical injury.
Surveillance: For all individuals: physical examination at least every six months. Annual assessment of: pain severity; disease burden including quality of life and activities of daily living, endurance tests to evaluate functional status of the cardiovascular, pulmonary, musculoskeletal, and nervous systems; upper and lower extremities for functionality and malalignment, hips for dysplasia/subluxation, and thoracolumbar spine for kyphosis. Neurologic examination every six months to assess for spinal cord compression; yearly whole-spine MRI in neutral position and cervical spine flexion-extension MRI if the results are inconclusive; spine radiographs every two to three years. Annual assessment of heart rate; electrocardiogram and echocardiogram every one to three years depending on disease course. Polysomnography every three years to assess for obstructive sleep apnea; annual pulmonary function in children older than age five years until growth stops, then every two to three years. Monitor nutritional status using MPS IVA-specific growth charts. Perform vision and eye exam at least annually, dental evaluation every six to 12 months, and annual audiogram. For those on enzyme replacement therapy: annual assessment of pain, disease burden parameters, and pulmonary function tests; urine keratan sulfate or total glycosaminoglycans every six months.
Agents/circumstances to avoid: Excessive weight gain; beta-blockers.
Genetic counseling.
MPS IVA is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a GALNS pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the GALNS pathogenic variants have been identified in an affected family member, carrier testing for at-risk family members, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
Mucopolysaccharidosis IVA (MPS IVA) should be suspected in an individual with the following findings on medical history, physical examination, skeletal radiographs, ophthalmologic examination, and family history; and suggestive laboratory findings [Wood et al 2013].
Medical history
- The majority of affected individuals do not have distinctive clinical findings at birth. However, some features may be present at birth including prominent forehead, pectus carinatum, kyphosis, and abnormal spine radiograph (see Skeletal radiographs)
- History of adenoidectomy, tonsillectomy, hernia repair, ear ventilation tubes (general findings for all MPS disorders)
- History of cervical spine decompression and/or fusion or a history of surgery for limb alignments (unique to MPS IVA among all MPS disorders)
- Respiratory compromise (sleep apnea, endurance limitations, snoring)
- Cardiac valve abnormalities
- Dental abnormalities
Physical examination. In severe MPS IVA the following findings are usually observed between ages one and three years; in slowly progressive MPS IVA the following findings may not become evident until as late as the second decade of life:
- Marked disproportionate short stature with short trunk and normal limbs (arm span exceeds height)
- Gibbus (short-segment structural thoracolumbar kyphosis resulting in sharp angulation of the back), kyphosis, and scoliosis
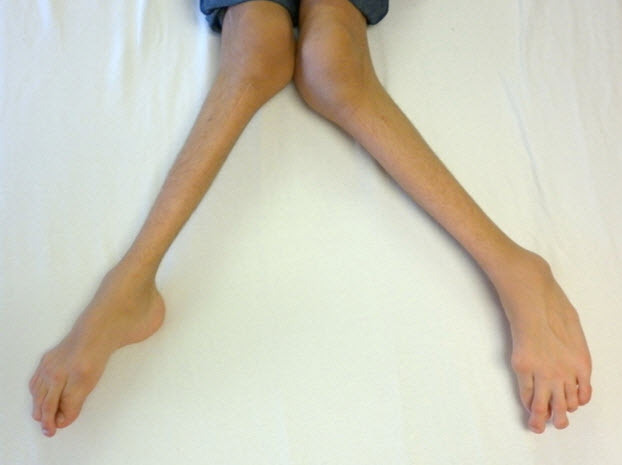
- Genu valgum (knock-knee) (Figure 5)
- Hypermobile joints
- Waddling gait with frequent falls

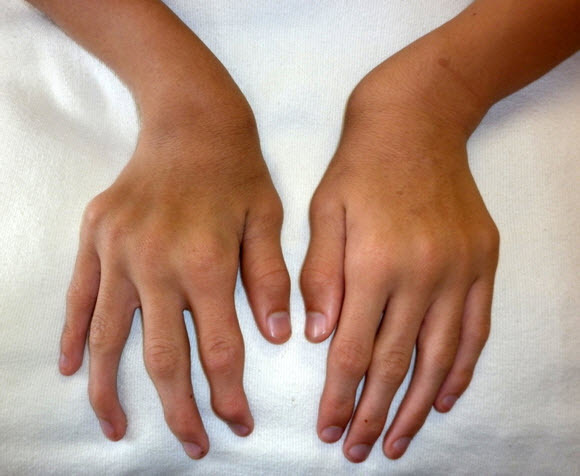
Figure 1.
Ulnar deviation of both wrists and joint enlargement in a male age 15 years with MPS IVA

Figure 2.
Shortened forearm and ulnar deviation of the wrist in a male age 15 years with MPS IVA

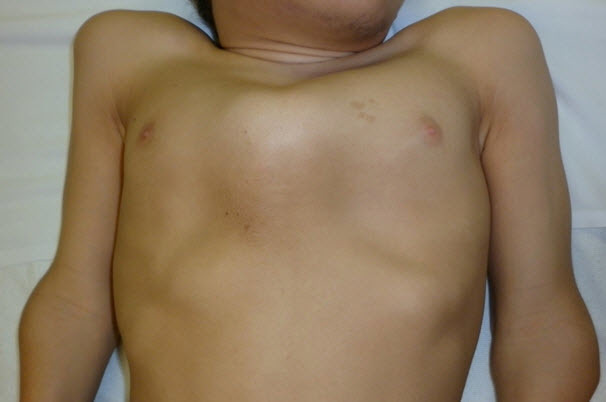
Figure 3.
Pectus anomaly and short neck in a male age 15 years with MPS IVA

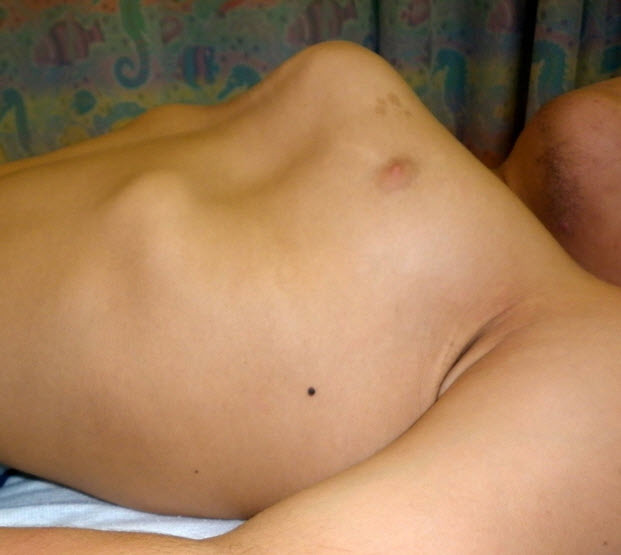
Figure 4.
Lateral view of chest showing severe pectus carinatum in a male age 15 years with MPS IVA

Figure 5.
Severe genu valgum (knock-knee) in a male age 15 years with MPS IVA
Skeletal radiographs
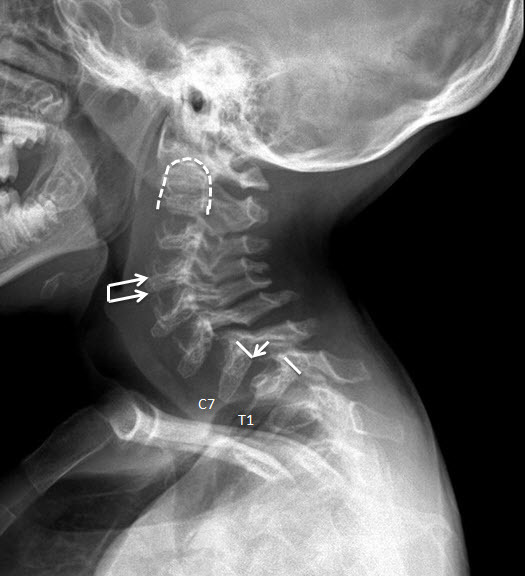
- Odontoid hypoplasia with subsequent cervical instability (Figure 6)
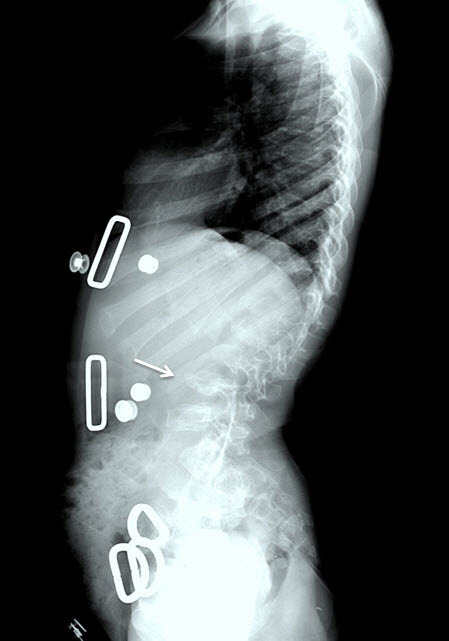
- Kyphosis (curving of the spine that causes a bowing or rounding of the back, which leads to a hunchback or slouching posture) (Figure 7)
- Gibbus (structural kyphosis) with wedging of one or more adjacent vertebrae (Figure 7)Note: The radiographic abnormalities of the lumbar spine can be detected at birth in infants with rapidly progressive disease [Tomatsu et al 2011].
- Scoliosis
- Pectus carinatum or (less frequently) excavatum
- Short ulnas, ulnar deviation of the radial epiphysis, and delayed bone maturation
- Short metacarpals with the proximal ends of the second to fifth metacarpals rounded or pointed [White et al 2014]
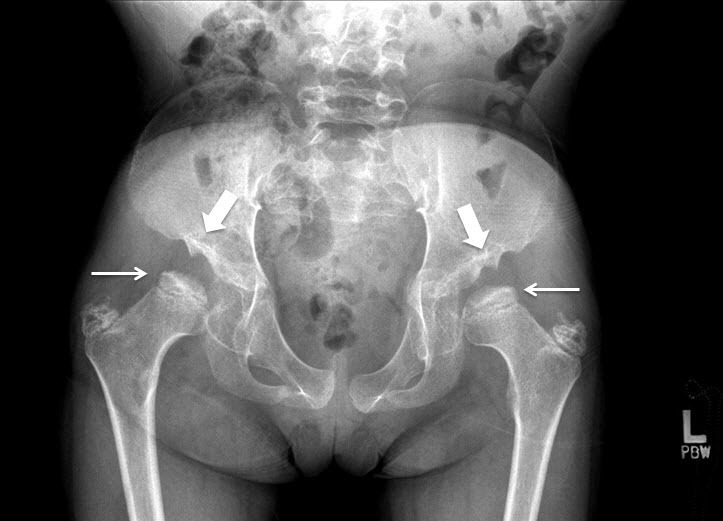
- Flared iliac wings, flattening of femoral epiphyses (Figure 8), and coxa valga
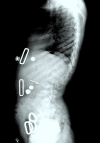
Figure 7.
Lateral spine radiograph of a female age eight years with MPS IVA. Note platyspondyly (flattened vertebrae) with anterior beaking (arrow).

Figure 8.
Hip radiograph of a female age eight years with MPS IVA. Note bilateral irregular flattening of the capital femoral epiphyses (thin arrows), irregular dysplastic acetabuli with lateral joint subluxation (thick arrows).
Note: Skeletal abnormalities are observed before physical abnormalities [Montaño et al 2007, Tomatsu et al 2011].
Ophthalmologic examination. Corneal clouding, astigmatism, cataracts, punctate lens opacities, open-angle glaucoma, optic disc swelling, optic atrophy, and/or retinopathy. Visual impairment can be secondary to several factors [Peracha et al 2018].
Family history consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.
Suggestive laboratory findings
- Qualitative urine glycosaminoglycan (GAG) analysis, which uses thin layer chromatography or electrophoresis to identify specific types of GAGs [Wood et al 2013], demonstrates keratan sulfate (KS) and chondroitin 6-sulfate (C6S).Note: The presence of KS (on qualitative analysis) with or without abnormal quantitative urine GAGs has been observed in some affected individuals.
- Quantitative urine GAG analysis, which measures the total amount of GAGs, urine KS, and C6S by ELISA method or LC-MS/MS:
- Elevated KS indicates deficiency of either the enzyme N-acetylgalactosamine 6-sulfatase (in MPS IVA) or the enzyme B-galactosidase (in MPS IVB);Note: Urine KS levels in younger individuals are higher than in older individuals due to the decrease in cartilage formation in older persons. The urine KS levels in individuals with MPS IVA and healthy controls were highest at age one to five years and declined with age; the levels reached a plateau after age 20 years [Sawamoto et al 2020].
- Elevated C6S indicates deficiency of the enzyme N-acetylgalactosamine 6-sulfatase (MPS IVA).
- Both qualitative and quantitative urine GAGs can be normal in some affected individuals. Thus, further enzymatic or molecular evaluation of a child with clinical evidence of MPS IV is warranted even when GAG analysis is normal.
Establishing the Diagnosis
The diagnosis of MPS IVA is established in a proband with: (1) low N-acetylgalactosamine 6-sulfatase (GALNS) enzyme activity in cultured fibroblasts or leukocytes OR (2) biallelic pathogenic (or likely pathogenic) variants in GALNS identified on molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include any likely pathogenic variants. (2) Identification of biallelic GALNS variants of uncertain significance (or of one known GALNS pathogenic variant and one GALNS variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of GALNS is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications and deep intronic variants may not be detected. If only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
- A multigene panel that includes GALNS and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Table 1.
Molecular Genetic Testing Used in Mucopolysaccharidosis Type IVA
N-acetylgalactosamine 6-sulfatase (GALNS) enzyme activity can be used when:
- Clinical findings strongly indicate MPS IVA and urine GAG analysis is normal; AND/OR
- Molecular genetic testing fails to identify biallelic pathogenic variants in GALNS.
In addition, establishing a diagnosis of MPS IVA by enzyme analysis may aid in interpretation of sequencing variants of uncertain significance. GALNS enzyme activity can be measured in cultured fibroblasts or leukocytes. Because each laboratory has its own normal range of enzyme activity, results from different laboratories cannot be directly compared. The level of residual enzyme activity may correlate with disease severity.
Note: (1) Low enzyme activity can be caused by other disorders including:
- Multiple sulfatase deficiency (MSD), deficient activity of several sulfatases (including GALNS). Thus, when GALNS enzyme activity is abnormal, additional sulfatase enzymes need to be assayed to evaluate for MSD.
- Mucolipidosis II and mucolipidosis III (see GNPTAB-Related Disorders, ML III Gamma). Mucolipidosis II and mucolipidosis III impair mannose-6-phosphate lysosomal enzyme targeting, leading to low lysosomal enzyme activity in fibroblasts, high lysosomal enzyme activity in plasma, and relatively unchanged lysosomal enzyme activity in leukocytes.
(2) Because the clinical manifestations of MPS IVA and MPS IVB are indistinguishable, it is customary to measure B-galactosidase enzyme activity at the same time.
Clinical Characteristics
Clinical Description
Mucopolysaccharidosis type IVA (MPS IVA) comprises a clinical continuum ranging from a severe and rapidly progressive form to a slowly progressive form. In the past, the two forms were distinguished by height, the subjective assessment of severity of bone deformity, and survival [Tomatsu et al 2011]; however, neither clinical nor biochemical findings can provide clear distinctions between the two forms and, thus, the MPS IVA phenotype should be considered a continuum from severe to slowly progressive.
Some features including prominent forehead, pectus carinatum, kyphosis, and abnormal spine radiograph may be detected at birth [Peracha et al 2018]. However, the majority of infants have no distinctive clinical findings. The severe form is usually apparent between ages one and three years. The slowly progressive form may not become evident until late childhood or adolescence [Montaño et al 2007].
In both the severe form and slowly progressive form, the initial presentations vary and individuals may present with only a single finding or several findings. Kyphoscoliosis, genu valgum (Figure 5), and pectus carinatum (Figure 3 and Figure 4) are the most common initial manifestations of the severe form [Montaño et al 2007]. In contrast, hip problems including pain and stiffness (due to collapse and flattening of the proximal femoral epiphysis) are common initial manifestations of the slowly progressive form [Hecht et al 1984, Wraith 1995, White et al 2014].
Because descriptions of the natural history of MPS IV published in the past may not have distinguished between MPS IVA (Morquio syndrome type A; accounting for >95% of affected individuals) and MPS IVB (Morquio syndrome type B; <5% of affected individuals), the following information is relevant to both MPS IVA and MPS IVB.
While the skeletal findings of MPS IVA are the hallmark findings, involvement of other organ systems can lead to significant morbidity, including respiratory compromise, obstructive sleep apnea, valvular heart disease, hearing impairment, corneal clouding, dental abnormalities, and hepatomegaly. Compression of the spinal cord results in neurologic involvement especially when disease is recognized later in life [Neufeld & Muenzer 2001, Tomatsu et al 2011, Solanki et al 2013].
Coarse facial features are also present but milder than in other mucopolysaccharidoses (see Differential Diagnosis).
Children with MPS IVA typically have normal intellectual ability.
Ligamentous laxity and joint hypermobility are distinctive features of MPS IVA, and are rare among other storage disorders.
Musculoskeletal
Skeletal findings worsen over time. The combination of bone and joint involvement leads to pain and arthritis that result in subsequent disability.
Upper-extremity involvement is also progressive and can impair hand-wrist strength and limit ability to perform some activities of daily living, such as using a fork. Hypermobility and ulnar deviation of the wrist joint due to premature cessation of ulna growth are distinctive features of MPS IVA.
Lower-extremity involvement, which is universal and progressive if untreated [Holzgreve et al 1981, Dhawale et al 2012], generally consists of malalignment due to progressive hip subluxation and/or valgus deformity. It can lead to significant gait alteration; hip, knee, and/or ankle pain with activity; and decreased endurance [Dhawale et al 2012, Peracha et al 2018].
Knee and ankle valgus are the most common lower-extremity deformities. Genu valgum (knock-knee) results from distal femoral and proximal tibial involvement and laxity of the collateral ligament.
A longitudinal study using the Pediatric Evaluation of Disability Inventory and the Functional Independence Measure found severely limited joint mobility in persons with MPS IVA, generally with loss of ambulation late in the disease course. Aggressive and long-term intervention by a team of physical therapists and rehabilitative specialists is often needed to optimize mobility (see Management) [Guarany et al 2012].
Hip dysplasia. Early in the disease course, the capital femoral epiphyses are small and the acetabula are shallow. Subsequent progressive flattening and fragmentation of the capital femoral epiphyses in the femoral head and acetabular dysplasia result in hip dislocation, arthritis, and severe joint restriction, causing affected individuals to become wheelchair bound [Tomatsu et al 2011, Peracha et al 2018].
Spinal cord compression may occur in any spinal segment; cervical spinal compression is the most common site. Spinal cord compression can be caused by cervical instability, unossified fibrocartilage associated with an abnormal odontoid process, ligamentous laxity, cartilaginous and ligamentous hypertrophy at the atlantoaxial joint, glycosaminoglycan (GAG) deposition in the extradural space, disc protrusion, thoracolumbar kyphosis, and acquired central canal stenosis [Lipson 1977, Ransford et al 1996, Tomatsu et al 2011, McKay et al 2012, Solanki et al 2013].
Odontoid hypoplasia leading to atlantoaxial instability, which later may result in upper cervical spinal cord compression, occurs in 90% of affected individuals (Figure 6) [Hughes et al 1997]. Persons with untreated atlantoaxial instability often do not survive beyond the second or third decade because minor falls and/or neck extension can result in quadriparesis or sudden death [Tomatsu et al 2011].
Spinal canal stenosis may be diffuse or focal. The causes of spinal canal stenosis are similar to the causes of spinal cord compression and include: kyphosis, disc protrusion, generalized thickening of posterior longitudinal ligament, or localized thickening of intervertebral ligaments due to GAG deposition.
Lumbar spine malalignment (i.e., thoracolumbar kyphosis) in older individuals can result in focal spinal stenosis, compressive myelopathy, and paraplegia [Dalvie et al 2001].
Ligamentous laxity resulting in hypermobile joints is common; however, decreased joint mobility can be observed in the large joints including knees, hips, and elbows [Neufeld & Muenzer 2001].
Neurologic
At the time of diagnosis, individuals typically have normal developmental milestones and normal intellectual ability. The neurologic findings of MPS IVA are most often secondary to spinal abnormalities in the neck and/or lumbar region. The increased risk for neurologic compromise makes developmental delay and learning disabilities more common in children with MPS IVA than in unaffected children [Montaño et al 2007].
Subtle abnormal brain MRI findings such as prominent perivascular space, enlarged lateral ventricles, and prominent frontal CSF were reported in eight of 14 individuals with MPS IVA [Davison et al 2013]. From the same study, neurocognitive evaluation revealed behavioral issues including anxiety, depression, decreased attention span, and somatic complaints. Whether these findings are caused by disease-specific biochemical abnormalities, chronic illness, or a combination of the two is unknown.
Cardiac
Cardiac complications include ventricular hypertrophy and early-onset severe valvular involvement. Coronary intimal sclerosis has also been reported [Hendriksz et al 2013].
In a multicenter, multinational, cross-sectional study (MorCAP) involving 325 individuals with MPS IVA, valvular regurgitation was more common than valvular stenosis. Among those with valvular regurgitation, tricuspid regurgitation was the most common (35%). Mitral regurgitation, aortic regurgitation, and pulmonary regurgitation were found in 25%, 19%, and 14% of affected individuals, respectively [Harmatz et al 2013]. Individuals with MPS IVA usually have an abnormally age-related aortic root dilatation, high heart rate and high myocardial index to compensate for small left ventricular diameter, impaired diastolic filling, and low stroke volume [Hendriksz et al 2015, Akyol et al 2019].
Respiratory
Respiratory complications are a major cause of morbidity and mortality. Airway obstruction, sleep-disordered breathing, and restrictive lung disease have been described.
GAG accumulation in the adenoids, tonsils, pharynx, larynx, trachea, and bronchial tree leads to adenotonsillar hypertrophy, tracheal distortion, tracheo- and bronchomalacia, and obstructive sleep apnea [Semenza & Pyeritz 1988, Walker et al 2003]. Deposition of GAGs in the trachea and bronchi could cause tortuosity of the airway leading to buckling and airway obstruction when the neck is flexed. If not recognized (particularly during cervical spine fusion/stabilization surgery) and if the head and neck are fused in a flexed position, acute obstruction may result in tracheal extubation [Solanki et al 2013]. In addition to tracheal narrowing from GAG accumulation, external factors including brachiocephalic artery crossing the anterior of the trachea and small thoracic inlet also contribute to tracheal stenosis [Peracha et al 2018].
Because of atlantoaxial instability and upper-airway obstruction, persons with MPS IVA prefer to sleep prone on a flat surface without a pillow in order to keep the neck extended and minimize the tortuosity of the airway.
Restrictive lung disease results from a small thorax, chest wall anomalies, spine deformities, neuromuscular compromise from cervical myelopathy, and hepatomegaly causing upward displacement of the diaphragm [Walker et al 2003, Hendriksz et al 2013].
If respiratory complications are not recognized or are not treated, cor pulmonale and respiratory failure can ensue, leading to early death [Hendriksz et al 2013, Peracha et al 2018].
Growth
Growth and final height are used as an indicator of severe phenotype. Children with MPS IVA have a normal birth weight and a longer-than-normal birth length. The growth velocity decreases between ages one and three years and growth stops around ages seven to eight years [Montaño et al 2007, Peracha et al 2018]. By age 18 years, the average height in males is 123 cm and in females 117 cm, compared to 177 cm and 163 cm in unaffected males and females, respectively.
Eye
Ophthalmologic findings are present in more than 50% of individuals with MPS IVA. Natural history studies have not been performed; thus, it is not possible to predict the age of onset of ophthalmologic findings [Wood et al 2013].
Slowly progressive corneal clouding, found in 50% of affected individuals (ages 1-65 years), is the most common ophthalmologic finding in MPS IVA.
Other less common ophthalmologic findings include: astigmatism, cataracts, punctate lens opacities, open-angle glaucoma, optic disc swelling, optic atrophy, and retinopathy. While rare, these ophthalmologic findings can be serious secondary complications [Hendriksz et al 2013, Hendriksz et al 2015].
Pseudoexophthalmos, the appearance of a bulging eye secondary to a shallow orbit, can cause exposure keratitis and also be of cosmetic concern.
Dental
Deciduous teeth erupt normally and are widely spaced and discolored with thin irregular (stippled) enamel and small pointed cusps which flatten over time with normal wear.
Permanent teeth also have hypoplastic enamel, and are widely spaced with flared upper incisors [Onçağ et al 2006, Peracha et al 2018].
Hearing
Mild-to-moderate hearing loss is common in individuals with MPS IVA. Hearing impairment is often noted toward the end of the first decade.
Mixed (i.e., combined conductive and sensorineural) hearing loss is more common than conductive or sensorineural hearing loss alone.
Conductive hearing loss is secondary to recurrent middle ear infections, serous otitis media, and deformity of the ossicles [Hendriksz et al 2013].
Sensorineural hearing loss secondary to GAG accumulation in the inner ear and/or central nervous system has been described [Ruckenstein et al 1991, Simmons et al 2005, Hendriksz et al 2013].
Genotype-Phenotype Correlations
Genetic alterations in GALNS that are predicted to severely affect protein function, such as deletions and nonsense variants, are common in individuals with rapidly progressive growth failure [Montaño et al 2007, Morrone et al 2014].
The location of variants in the tertiary GALNS protein structure may determine the severity of the phenotype. Variants that interfere with the salt bridge, alter the active site, or destroy the hydrophobic core contribute to a severe phenotype, whereas variants located on the surface of the protein (e.g., p.Asp60Asn, p.Asn204Lys, p.Arg259Gln) likely contribute to a milder phenotype [Sawamoto et al 2020].
Several variants have been reported in individuals with severe phenotypes; these include c.29G>A (p.Trp10Ter), c.1519T>C (p.Cys507Arg), c.1520 G>T (p.Cys507Phe) [Zanetti et al 2019].
Nomenclature
Much of the older literature and more complete natural history studies were performed prior to understanding of the basis of MPS IVA (N-acetylgalactosamine 6-sulfatase deficiency or biallelic GALNS pathogenic variants) and MPS IVB (B-galactosidase deficiency or biallelic GLB1 pathogenic variants). Thus, the term MPS IV refers to individuals with a clinical diagnosis without an enzymatic and/or molecular diagnosis, whereas the more specific terms MPS IVA and MPS IVB refer to individuals with an enzymatic and/or molecular diagnosis.
Mucopolysaccharidosis type IVA (MPS IVA), also known as Morquio syndrome type A, was initially characterized by Morquio [1929] and Brailsford [1929].
MPS IVA and MPS IVB are known as Morquio syndrome type A and type B, respectively.
Prevalence
MPS IVA is rare. The prevalence in Australia has been estimated at 1:926,000, and in the UK at 1:599,000. The birth prevalence for MPS IVA ranges from 1:71,000 to 1:179,000 across multiple countries [Leadley et al 2014]. In one German study, the incidence of MPS IVA was 1:270,000 [Baehner et al 2005]. Similarly, the incidence of MPS IVA in Italy was estimated at 1:300,000 live births [Caciotti et al 2015].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are associated with germline pathogenic variants in GALNS.
Differential Diagnosis
Mucopolysaccharidosis type IVB (MPS IVB; see GLB1-Related Disorders) and mucopolysaccharidosis IVA (MPS IVA) are clinically indistinguishable. In MPS IVB, the accumulation of keratan sulfate occurs due to biallelic pathogenic variants in GLB1, the gene encoding the enzyme B-galactosidase. Of individuals with the MPS IV phenotype, MPS IVA accounts for more than 95% of affected individuals and MPS IVB accounts for fewer than 5% of affected individuals. In most individuals with clinical findings of MPS IV, MPS IVA can be distinguished from MPS IVB only by biochemical testing and/or molecular genetic testing.
Although novel GLB1 pathogenic variants identified in MPS IVB often map to the substrate binding regions of the protein [Ohto et al 2012], some pathogenic variants are associated with both GM1 gangliosidosis and MPS IVB.
Note: Biallelic pathogenic variants in GLB1 also cause GM1 gangliosidosis, a lysosomal storage disease with severe neurologic outcomes and skeletal dysplasia. The spectrum of GM1 gangliosidosis comprises a continuum of infantile, late-infantile, juvenile, and adult forms. The infantile form often results in death by age two years, while life span may be normal in the adult form.
Other mucopolysaccharidoses are summarized in Table 2.
Table 2.
Other Mucopolysaccharidoses to Consider in the Differential Diagnosis of Mucopolysaccharidosis IVA
Spondyloepiphyseal dysplasia (SED) has similar radiographic findings. Clinical features, especially extraskeletal features, could distinguish SED from MPS IVA. See Schimke Immunoosseous Dysplasia and X-Linked Spondyloepiphyseal Dysplasia Tarda.
Legg-Calve-Perthes disease (OMIM 150600). In some instances mild MPS IVA can manifest only with hip pain at the onset of disease, which can lead to an initial misdiagnosis of Legg-Calve-Perthes disease.
Note: COL2A1 pathogenic variants have been associated with a Legg-Calve-Perthes-like phenotype (more accurately dysplastic proximal femoral epiphyses). Bilateral hip involvement, especially symmetric and synchronous, is suggestive of a type II collagen disorder. (See Type II Collagen Disorders Overview.)
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with mucopolysaccharidosis type IVA (MPS IVA), the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended [Hendriksz et al 2013, Solanki et al 2013, Hendriksz et al 2015, Peracha et al 2018, Akyol et al 2019].
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with Mucopolysaccharidosis IVA
Treatment of Manifestations
Management of individuals with MPS IVA is best undertaken by the following multiple specialists, coordinated by a physician specializing in the care of individuals with complex medical problems:
- Physiatrist (specialist in physical medicine and rehabilitation) to optimize mobility and autonomy
- Physical therapist to optimize mobility
- Occupational therapist to optimize autonomy
- Psychological support to optimize coping skills and quality of life
- Education professionals to optimize learning in a medically fragile individual
- Consideration of referral to family therapy to help normalize the experience for the affected individual, parents, sibs, and extended family members
- Home care for affected individuals with multiple medical equipment needs
- Hospice for end-of-life care
Enzyme Replacement Therapy (ERT)
Recombinant human GALNS ERT (elosulfase alfa, or Vimizim™) was approved by the FDA in February 2014.
- The recommendation dose is 2 mg/kg/week intravenous. Although the treatment with ERT is not curative, ERT could improve endurance and overall quality of life.
- Premedication (30-60 minutes prior to each enzyme infusion) with a non-sedating antihistamine (if possible) with or without antipyretics is recommended to prevent infusion-associated reactions.
- A Phase III clinical trial demonstrated a statistically significant improvement in the 6MWT distance in the 2 mg/kg weekly dose group compared to the placebo group. The three-minute stair climb test and respiratory function were improved with the treatment but the differences were not statistically significant.
- The long-term effects of this treatment on the skeletal features of MPS IVA are still unclear (see Therapies Under Investigation). The efficacy of ERT in improving pathology in the musculoskeletal system may be limited because of poor biodistribution of the enzyme in avascular tissue. At least two studies have reported no change in final height especially in individuals with a severe phenotype, even though ERT was started before age three years. Both treated and untreated individuals reached their final height around age ten years and there was no significant difference in growth velocity [Doherty et al 2019].
- The impact of ERT on mildly affected individuals after 52 weeks of treatment was minimal, lacking significant change on cardiac or pulmonary function.
Musculoskeletal
For published orthopedic management guidelines, see White et al [2014]. The level of physical activity should be monitored by specialists in orthopedic surgery, neurology, physiatry, and physical therapy to optimize mobility while preventing joint injury, joint malalignments, and cervical cord damage.
Upper extremities. Nonoperative interventions, such as external wrist splints, may be considered. Surgical intervention including partial or complete wrist fusion may be necessary to stabilize wrist range of motion.
Knee and ankle valgus. Lower-extremity malalignment associated with progressively poor mechanical alignment and decreasing endurance requires intervention; however, no absolute indications for intervention exist.
- Growth modulation, also called guided growth (temporary surgical tethering of the growth plate to allow gradual correction of the deformity) or realignment osteotomies have been successful [Dhawale et al 2012, White et al 2014]. When deformity is noted or the tibial-femoral angle is >15°, growth modulation should be considered [Akyol et al 2019].
- Early detection and evaluation may allow surgical tethering of the growth plate to treat mild-to-moderate lower-extremity angular deformities in children with open physes (growth plates). Typically this procedure is less invasive and allows for easier recovery than realignment osteotomies.
- Once the growth plates close, distal femoral and proximal tibial osteotomies are needed to acutely or gradually (with the use of external fixators) correct lower-extremity malalignment.
- Ankle malalignment is often corrected by a distal tibial osteotomy with distal tibial screw hemiepiphysiodesis [Tomatsu et al 2011].
Hip dysplasia. Surgery can manage pain and alignment and enhance mobility.
- Hip reconstruction includes either femoral or acetabular osteotomy for mild cases or combined acetabular and femoral osteotomy for severe cases. Augmentation of acetabular bone stock and customized implants by using cortical grafts from the inner table of the ilium are usually required due to a shallow acetabulum [White et al 2014].
- Total hip arthroplasty may be required in young adults experiencing significant hip pain which cannot be corrected by reconstructive techniques.
Odontoid hypoplasia. When upper cervical spine instability is documented or when clinical findings of cervical myelopathy are present, occipito-cervical or upper cervical decompression and fusion are required to stabilize the upper cervical spine and relieve cervical cord compression.
- To minimize neurologic injury and maximize function, intervention in children is recommended when radiographic signs of cervical compression are present, even in the absence of symptoms.
- Affected individuals undergoing surgical fusion typically do well; minor secondary complications can include pin site infections, pressure sores, and long-term difficulty with endotracheal intubation.
- Note: It is important for clinicians to be aware that cervical myelopathy from upper cervical instability may result in deteriorating endurance and worsening gait. If myelopathy is suspected, obtain cervical spine radiographs and MRI (see Surveillance). The affected individual should be referred for evaluation by a pediatric orthopedic surgeon or neurosurgeon at a tertiary care facility.
Lumbar spine malalignment. Thoracolumbar kyphosis (resulting from vertebral hypoplasia) may be progressive and symptomatic.
- When kyphosis is <45°, the risk of progressive deformity is less than with a greater curve, but warrants clinical and radiographic monitoring.
- When kyphosis is >45°, progression is likely. Although extensive bracing with an orthosis or a cast does not prevent progression of the thoracolumbar kyphosis, it may delay the need for surgical intervention during a period of growth and development.
- Anterior and posterior circumferential spinal fusion are indicated if one or more of the following are present:
- Progressive thoracolumbar kyphosis >70°
- Uncontrolled back pain
- Neurologic changes related to spinal stenosis
Spinal cord function monitoring during surgery. Because subacute spinal stenosis and/or dynamic spinal stenosis could lead to spinal cord injury, procedures involving cervical spine manipulation, prone positioning (including spinal surgery), and/or prolonged time under anesthesia (e.g., exceeding 45 minutes), should be considered for intraoperative neurophysiologic monitoring (IONM). IONM uses somatosensory and motor evoked potentials (SSEPs or MEPs) to monitor spinal cord function [Solanki et al 2013, Walker et al 2013]. Note: While spinal infarct during surgery was reported in a few individuals with skeletal dysplasia [Tong et al 2012, Pruszczynski et al 2015], data demonstrating consistent improvement of outcome with this monitoring technology are limited [Solanki et al 2013].
Cardiac
Elevated heart rates could indicate a compensation mechanism, secondary to small left ventricular diameter and small stroke volume; thus tachycardia treatment with beta-blockers should be avoided. Valve replacement may be considered for symptomatic individuals with progressive valvular problems [Hendriksz et al 2015, Akyol et al 2019]. Risks need to be carefully weighed for valve replacement, either mechanical (lifelong use of anticoagulants) or bioprosthesis (increased risk of valve dysplasia, degradation, and calcification).
Bacterial endocarditis prophylaxis is recommended for those at high risk, including those with a prosthetic cardiac valve, prosthetic material used for cardiac valve repair, or previous infective endocarditis [Wilson et al 2007].
Respiratory
Upper-airway obstruction and obstructive sleep apnea are managed by removal of enlarged tonsils and adenoids at an average age of seven years [Montaño et al 2007]. Note: Even with this intervention, the rate of obstructive sleep apnea in children with a mucopolysaccharidosis is much higher than in the general population; therefore, prompt clinical evaluation and referral for polysomnography are appropriate [Nashed et al 2009].
In persons with diffuse narrowing of the airway in whom adenotonsillectomy only temporally relieves upper-airway obstruction, other interventions to consider are: CPAP (continuous positive airway pressure), NIPPV (noninvasive positive pressure ventilation), tracheostomy, and possibly tracheal reconstructive surgery.
Lower-airway obstruction manifest as wheezing and recurrent infection is managed by inhaled and/or oral bronchodilators and, in some instances, corticosteroids.
Restrictive lung disease is managed by supportive treatment.
Due to increased risk for pulmonary infection, all affected individuals should receive influenza and pneumococcal immunizations as well as routine immunizations.
Anesthesia. Procedures requiring anesthesia require considerable planning and are best performed in a facility in which anesthesiologists are experienced with the airway issues of MPS IVA, such as abnormal anatomy and glycosaminoglycan (GAG) accumulation, unstable cervical spine, and progressive pulmonary disease (both restrictive and obstructive). Theroux et al [2012], who published the largest cohort of children with MPS IV undergoing anesthesia, made specific recommendations for care during anesthesia.
Preoperative assessment should include history of response to anesthesia and any evidence of airway obstruction, cardiac evaluation including electrocardiogram and echocardiography, evaluation of respiratory function (spirometry and polysomnography), and airway fluoroscopy [Muhlebach et al 2011, Tomatsu et al 2011].
Endotracheal intubation likely includes use of a video laryngoscope, fiberoptic bronchoscope with or without a laryngeal mask airway (LMA), a smaller endotracheal tube than expected for age and/or size, and use of LMA for short procedures. These techniques help maintain a neutral neck position. Although nasal intubation is an option, GAG deposits can lead to narrowing of the nasal passages and increased propensity to bleeding [Aziz et al 2011, Muhlebach et al 2011, Walker et al 2013, Akyol et al 2019].
Intraoperative neurophysiologic monitoring (see Spinal cord function monitoring during surgery) during all spinal surgeries and long, complicated procedures is strongly recommended. Intrathecal and epidural techniques should be avoided. During operation, steroid prophylaxis to reduce airway edema is suggested [Akyol et al 2019].
Postoperative narcotic management should be judicious; multimodal analgesics and non-narcotic medications are preferable to avoid exacerbating preexisting respiratory issues, such as sleep apnea.
Postoperative complications including pulmonary edema have been described [Morgan et al 2002].
Due to risk of postoperative complications, some individuals may require close monitoring for at least 24-48 hours.
Growth
Height of children with MPS IVA is best plotted on growth charts specific for MPS IVA [Montaño et al 2007, Montaño et al 2008].
Nutrition should be optimized with a balanced diet and adequate vitamin D and calcium to assure bone health.
Eye
Corneal opacification often causes reduced vision in early childhood, necessitating corneal transplantation (deep lamellar keratoplasty or penetrating keratoplasty), for which the outcome can vary. Other factors which may cause reduced vision such as retinopathy should be excluded before considering corneal transplantation. Recurrence of opacities within the first year post keratoplasty has been reported, making this a temporary measure for improving quality of life [Bothun et al 2011]. In addition, other ophthalmologic problems including glaucoma and retinopathy may limit the success of corneal transplantation. Cataract surgery may benefit those with cataracts.
Dental
Daily oral hygiene care, fissure sealing, and adequate fluoride supplementation help prevent cavities. Orthodontic management to correct malocclusion may be necessary.
Hearing
Because ventilation tube placement can minimize the long-term scarring associated with chronic middle-ear effusions and recurrent acute otitis media, and improve hearing in the long term, most children have ventilation tubes placed during the preschool years. At the first occasion, a long-lasting tympanostomy tube is recommended due to high risk of recurrent middle-ear effusion and the risk associated with sedation in individuals with MPS IVA [Hendriksz et al 2015].
The progressive hearing impairment observed in most individuals with MPS IVA benefits from hearing aids.
Learning Environment
Despite some physical limitation, individuals with MPS IVA have normal intellect and can thrive in an environment with academic and social stimulation. Children routinely attend regular class/school with assistance to prevent physical injury.
Surveillance
Individuals on ERT
The following should be assessed before and after initiation of ERT to determine treatment efficacy:
- Annual pain severity and disease burden assessments including quality of life and activities of daily living (See All Individuals.)
- Annual pulmonary function tests, including maximum voluntary ventilation (MVV) and forced vital capacity (FVC)
- Urine KS / urine GAG levels at baseline, then every six months
Note: The benefit of monitoring anti-elosulfase alfa antibodies is unknown.
All Individuals
Assessment of quality of life, disease burden, and endurance
- Every 6-12 months, track growth, pubertal stage, and progress; optimize ambulation.
- Annually, assess pain severity and disease burden including quality of life and activities of daily living.
- Annually, before and after surgical procedures, or as clinically indicated, perform endurance tests including six-minute walk test or timed 25-foot walk test to evaluate functional status of the cardiovascular, pulmonary, musculoskeletal, and nervous systems. Respiratory rate, pulse oximeter, and heart rate should be measured before and after the annual testing.
Musculoskeletal. White et al [2014] recommend the following guidelines for monitoring musculoskeletal involvement in those with MPS IVA (full text):
- Upper and lower extremities. Evaluate severity and progression of upper- and lower-extremity involvement at least annually.
- Evaluation of range of motion, grip and pinch strength, and functional assessments (e.g., functional dexterity test) of the upper extremities
- Assessment of lower-extremity alignment, including standing AP radiographs (as clinically indicated) and AP and frog leg lateral radiographs of the pelvis to assess hip dysplasia/subluxation when clinically indicated
- Spine. Neurologic examination every six months to assess for spinal cord compression [Solanki et al 2013]:
- For children who are reliable historians, at each clinic visit obtain a history of exercise tolerance and symptoms of myelopathy (e.g., extremity weakness; clumsiness; unsteady, changing gait; bowel or bladder dysfunction or lower back/leg pain).
- In those with multisegmental myelopathy, SSEPs and MEPs (if available) may provide detailed information.
- Plain radiographs of the cervical spine (AP, lateral, neutral, and flexion-extension) every two to three years
- Plain radiographs of the spine (AP and lateral views for the thoracolumbar spine) every two to three years
- MRI of the whole spine (neutral position)* annually and flexion-extension MRI of the cervical spine if results are inconclusive* Neutral, flexion, and extension lateral radiographs of the cervical spine should be obtained prior to cervical MRI to assess for atlantooccipital instability.
Cardiac. Evaluate heart rate annually; perform electrocardiogram and echocardiogram every one to three years depending on disease course [Braunlin et al 2011, Hendriksz et al 2013, Hendriksz et al 2015].
Respiratory
- For obstructive sleep apnea, annual history focused on sleep patterns and sounds. Evaluation by an otolaryngologist for adenotonsillectomy. Annual in-home screening sleep studies (which monitor oxygen saturation). Polysomnography every three years.
- To assess pulmonary function, annual MVV and FVC in children older than age five years until growth stops, then every two to three years. The benefit of noninvasive pulmonary function tests, impulse oscillometry, and thoracoabdominal motion analysis has been demonstrated in children with MPS IV [Rodriguez et al 2010, Akyol et al 2019].
- Fiberoptic examination at least annually or as clinically indicated [Akyol et al 2019]
Growth. Use MPS IVA-specific growth charts to monitor nutritional status [Montaño et al 2007, Montaño et al 2008]. Length/height and weight should be measured at every visit.
Eye
- Monitor for vision and ocular abnormalities with ophthalmologist examination at least annually.
- For those with rod and cone retinal dystrophy, perform retinal examination and electroretinography under scotopic and photopic conditions every five years [Hendriksz et al 2013].
Dental. Evaluate every six to 12 months.
Hearing. Perform yearly audiogram.
Agents/Circumstances to Avoid
Because excessive weight gain causes undue stress on the axial skeleton and may decrease the duration of independent ambulation, it is important to optimize nutrition for growth while maintaining a lean habitus.
Due to small ventricular diameter and stroke volume, beta-blockers should be avoided in the treatment of tachycardia.
Evaluation of Relatives at Risk
It is appropriate to evaluate apparently asymptomatic younger sibs of a proband in order to identify as early as possible those who would benefit from initiation of ERT (see Treatment of Manifestations). Evaluations can include:
- Molecular genetic testing if the pathogenic variants in the family are known;
- Analysis of N-acetylgalactosamine 6-sulfatase (GALNS) enzyme activity if the pathogenic variants in the family are not known.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
The experience of hematopoietic stem cell therapy is limited and has not been well studied. Gene therapy and substrate degradation enzyme therapy are in preclinical trials [Sawamoto et al 2020].
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Mucopolysaccharidosis type IVA (MPS IVA) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., presumed to be carriers of one GALNS pathogenic variant based on family history).
- Accurate recurrence risk counseling relies on carrier testing of both parents to confirm that they are both heterozygous for a GALNS pathogenic variant. If carrier testing detects a pathogenic variant in only one parent:
- And the child appears to have homozygous GALNS pathogenic variants, possible explanations include a large deletion on one allele (if not previously tested for) [Caciotti et al 2015] and uniparental isodisomy for chromosome 16 [Catarzi et al 2012];
- And the child has compound heterozygous GALNS pathogenic variants, the child may have one inherited pathogenic variant and one de novo pathogenic variant. (De novo variants are known to occur at a low but appreciable rate in autosomal recessive disorders [Jónsson et al 2017].)
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for a GALNS pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. The offspring of an individual with MPS IVA are obligate heterozygotes (carriers) for a GALNS pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a GALNS pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the GALNS pathogenic variants in the family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Once the GALNS pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing for MPS IVA are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Canadian Society for Mucopolysaccharide and Related DiseasesCanadaPhone: 800-667-1846Email: info@mpssociety.ca
- MedlinePlus
- Morquiosity
- MPS SocietyUnited KingdomPhone: 0345 389 9901Email: mps@mpssociety.org.uk
- National MPS SocietyPhone: 877-MPS-1001
- National Organization for Rare Disorders (NORD)Phone: 800-999-6673
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Mucopolysaccharidosis Type IVA : Genes and Databases
Table B.
OMIM Entries for Mucopolysaccharidosis Type IVA (View All in OMIM)
Pathophysiology
Mucopolysaccharidosis type IVA (MPS IVA) is caused by a deficiency of the lysosomal enzyme N-acetylgalactosamine-6-sulfatase (GALNS), which cleaves the keratan sulfate at the O-linked sulfate moiety of keratan sulfate (KS) and chondroitin-6-sulfate (C6S). The absence of the enzyme GALNS leads to intracellular accumulation of the glycosaminoglycans KS and C6S in the lysosomes of multiple tissues. The accumulation mainly in cornea and bone leads to the pathognomonic findings of corneal clouding and skeletal dysplasia [Neufeld & Muenzer 2001].
Missense, nonsense, splicing and deep intronic variants, as well as small deletions, small insertions, gross insertions/duplications, gross deletions, and complex rearrangements have been found in GALNS.
Mechanism of disease causation. Loss of function. The abnormal gene product demonstrates decreased enzymatic activity, leading to accumulation of sulfated intermediates primarily in the cartilage and its extracellular matrix. A GALNS pathogenic variant can lead to misfolding (and subsequent early degradation), mislocalization, or alterations in the catalytic domain either directly or through substrate binding sites [Rivera-Colón et al 2012].
Table 4.
Notable GALNS Pathogenic Variants
Chapter Notes
Author Notes
Dr Regier is the medical director in the Children's National Rare Disease Institute. Her main interest in in the field of Inborn Errors of Metabolism and educational outcomes research. She has been actively involved in clinical trials and implementation of new treatments for patients with Inborn Errors of Metabolism.
Dr Oetgen, an attending surgeon in the Department of Orthopaedics and Sports Medicine at Children's National Medical Center, has a special interest in the orthopaedic manifestations of genetic conditions affecting pediatric patients, including the mucopolysaccharidoses. He participates in the care of these patients in collaboration with the Children's National Hospital Department of Genetics.
Dr Tanpaiboon is a geneticist and researcher in the Division of Genetics and Metabolism at Children's National Rare Disease Institute. Her main interest is the field of Inborn Errors of Metabolism, particularly the lysosomal storage disorders (LSDs). She has been actively involved in international multicenter clinical trials of enzyme replacement therapy for MPS IVA and other LSDs.
Acknowledgments
Kathleen Crosby, MS, CGC and Lindsay Kehoe MS, CGC are gratefully acknowledged for providing excellent care for patients and their families
Revision History
- 17 June 2021 (sw) Comprehensive update posted live
- 24 March 2016 (ma) Comprehensive update posted live
- 11 July 2013 (me) Review posted live
- 18 January 2013 (pt) Original Submission
References
Literature Cited
- Akyol MU, Alden TD, Amartino H, Ashworth J, Belani K, Berger KI, Borgo A, Braunlin E, Eto Y, Gold JI, Jester A, Jones SA, Karsli C, Mackenzie W, Marinho DR, McFadyen A, McGill J, Mitchell JJ, Muenzer J, Okuyama T, Orchard PJ, Stevens B, Thomas S, Walker R, Wynn R, Giugliani R, Harmatz P, Hendriksz C, Scarpa M, et al. Recommendations for the management of MPS IVA: systematic evidence- and consensus-based guidance. Orphanet J Rare Dis. 2019;14:137. [PMC free article: PMC6567385] [PubMed: 31196221]
- Aziz MF, Healy D, Kheterpal S, Fu RF, Dillman D, Brambrink AM. Routine clinical practice effectiveness of the Glidescope in difficult airway management: an analysis of 2,004 Glidescope intubations, complications, and failures from two institutions. Anesthesiology. 2011;114:34-41. [PubMed: 21150569]
- Baehner F, Schmiedeskamp C, Krummenauer F, Miebach E, Bajbouj M, Whybra C, Kohlschütter A, Kampmann C, Beck M.Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis. 2005;28:1011-7. [PubMed: 16435194]
- Bothun ED, Decanini A, Summers CG, Orchard PJ, Tolar J. Outcome of penetrating keratoplasty for mucopolysaccharidoses. Arch Ophthalmol. 2011;129:138-44. [PubMed: 21320956]
- Brailsford J. Chondro-osteo-dystrophy: Roentgenographic anc clinical features of child with dislocation of vertebrae. Am J Surg. 1929;7:404–10. [PubMed: 770041]
- Braunlin EA, Harmatz PR, Scarpa M, Furlanetto B, Kampmann C, Loehr JP, Ponder KP, Roberts WC, Rosenfeld HM, Giugliani R. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34:1183-97. [PMC free article: PMC3228957] [PubMed: 21744090]
- Caciotti A, Tonin R, Mort M, Cooper DN, Gasperini S, Rigoldi M, Parini R, Deodato F, Taurisano R, Sibilio M, Parenti G, Guerrini R, Morrone A. Mis-splicing of the GALNS gene resulting from deep intronic mutations as a cause of Morquio a disease. BMC Med Genet. 2018;19:183. [PMC free article: PMC6180571] [PubMed: 30305043]
- Caciotti A, Tonin R, Rigoldi M, Ferri L, Catarzi S, Cavicchi C, Procopio E, Donati MA, Ficcadenti A, Fiumara A, Barone R, Garavelli L, Rocco MD, Filocamo M, Antuzzi D, Scarpa M, Mooney SD, Li B, Skouma A, Bianca S, Concolino D, Casalone R, Monti E, Pantaleo M, Giglio S, Guerrini R, Parini R, Morrone A. Optimizing the molecular diagnosis of GALNS: novel methods to define and characterize Morquio-A syndrome-associated mutations. Hum Mutat. 2015;36:357-68. [PubMed: 25545067]
- Catarzi S, Giunti L, Papadia F, Gabrielli O, Guerrini R, Donati MA, Genuardi M, Morrone A. Morquio A syndrome due to maternal uniparental isodisomy of the telomeric end of chromosome 16. Mol Genet Metab. 2012;105:438-42. [PubMed: 22178352]
- Dalvie S, Skinner J, Vellodi A, Noorden MH. Mobile thoracolumbar gibbus in Morquio type A: the cause of paraparesis and its management. J Pediatr Orthop B. 2001;10:328-30. [PubMed: 11727377]
- Davison JE, Kearney S, Horton J, Foster K, Peet AC, Hendriksz CJ.Intellectual and neurological functioning in Morquio syndrome (MPS IVa). J Inherit Metab Dis. 2013;36:323-8. [PubMed: 22231379]
- Dhawale AA, Thacker MM, Belthur MV, Rogers K, Bober MB, Mackenzie WG. The lower extremity in Morquio syndrome. J Pediatr Orthop. 2012;32:534-40. [PubMed: 22706472]
- Doherty C, Stapleton M, Piechnik M, Mason RW, Mackenzie WG, Yamaguchi S, Kobayashi H, Suzuki Y, Tomatsu S. Effect of enzyme replacement therapy on the growth of patients with Morquio A. J Hum Genet. 2019;64:625-35. [PubMed: 31019230]
- Guarany NR, Schwartz IV, Guarany FC, Giugliani R.Functional capacity evaluation of patients with mucopolysaccharidosis. J Pediatr Rehabil Med. 2012;5:37-46. [PubMed: 22543891]
- Harmatz P, Mengel KE, Giugliani R, Valayannopoulos V, Lin SP, Parini R, Guffon N, Burton BK, Hendriksz CJ, Mitchell J, Martins A, Jones S, Guelbert N, Vellodi A, Hollak C, Slasor P, Decker C. The Morquio A Clinical Assessment Program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab. 2013;109:54-61. [PubMed: 23452954]
- Hecht JT, Scott CI Jr, Smith TK, Williams JC. Mild manifestations of the Morquio syndrome. Am J Med Genet. 1984;18:369-71. [PubMed: 6431819]
- Hendriksz CJ, Al-Jawad M, Berger KI, Hawley SM, Lawrence R, Mc Ardle C, Summers CG, Wright E, Braunlin E. Clinical overview and treatment options for non-skeletal manifestations of mucopolysaccharidosis type IVA. J Inherit Metab Dis. 2013; 36:309-22. [PMC free article: PMC3590399] [PubMed: 22358740]
- Hendriksz CJ, Berger KI, Giugliani R, Harmatz P, Kampmann C, Mackenzie WG, Raiman J, Villarreal MS, Savarirayan R. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet A. 2015;167A:11-25. [PMC free article: PMC4309407] [PubMed: 25346323]
- Holzgreve W, Grobe H, von Figura K, Kresse H, Beck H, Mattei JF. Morquio syndrome: clinical findings in 11 patients with MPS IVA and 2 patients with MPS IVB. Hum Genet. 1981;57:360-5. [PubMed: 6793501]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389-97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Hughes DG, Chadderton RD, Cowie RA, Wraith JE, Jenkins JP. MRI of the brain and craniocervical junction in Morquio's disease. Neuroradiology.1997;39:381-5. [PubMed: 9189888]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519-22. [PubMed: 28959963]
- Leadley RM, Lang S, Misso K, Bekkering T, Ross J, Akiyama T, Fietz M, Giugliani R, Hendriksz CJ, Hock NL, McGill J, Olaye A, Jain M, Kleijnen J. A systematic review of the prevalence of Morquio A syndrome: challenges for study reporting in rare diseases Orphanet J Rare Dis. 2014;9:173. [PMC free article: PMC4251694] [PubMed: 25404155]
- Lipson SJ. Dysplasia of the odontoid process in Morquio's syndrome causing quadriparesis.J Bone Joint Surg Am. 1977;59:340-4. [PubMed: 403192]
- Mackenzie WG, Dhawale AA, Demczko MM, Ditro C, Rogers KJ, Bober MB, Campbell JW, Grissom LE. Flexion-extension cervical spine MRI in children with skeletal dysplasia: is it safe and effective? J Pediatr Orthop. 2013;33:91-8. [PubMed: 23232386]
- McKay SD, Al-Omari A, Tomlinson LA, Dormans JP. Review of cervical spine anomalies in genetic syndromes. Spine (Phila Pa 1976). 2012;37:E269-77. [PubMed: 22045003]
- Montaño AM, Tomatsu S, Brusius A, Smith M, Orii T. Growth charts for patients affected with Morquio A disease. Am J Med Genet A. 2008;146A:1286-95. [PubMed: 18412124]
- Montaño AM, Tomatsu S, Gottesman GS, Smith M, Orii T. International Morquio A Registry: clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis. 2007;30:165-74. [PubMed: 17347914]
- Morgan KA, Rehman MA, Schwartz RE. Morquio's syndrome and its anaesthetic considerations. Paediatr Anaesth. 2002;12:641-4. [PubMed: 12358664]
- Morquio L. Sur une forme de dystrophie. Bull Soc Pediatr (Paris). 1929;27:145-52.
- Morrone A, Caciotti A, Atwood R, Davidson K, Du C, Francis-Lyon P, Harmatz P, Mealiffe M, Mooney S, Oron TR, Ryles A, Zawadzki KA, Miller N. Morquio A syndrome-associated mutations: a review of alterations in the GALNS gene and a new locus-specific database. Hum Mutat. 2014;35:1271-9. [PMC free article: PMC4238747] [PubMed: 25137622]
- Muhlebach MS, Wooten W, Muenzer J. Respiratory manifestations in mucopolysaccharidoses. Paediatr Respir Rev. 2011;12:133-8. [PubMed: 21458742]
- Nashed A, Al-Saleh J, Gibbons J, MacLusky I, MacFarlane J, Riekstins A, Clarke J, Narange I. Sleep-related breathing in children with mucopolysaccharidosis. J Inherit Metab Dis. 2009;32:544-50 [PubMed: 19562504]
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, eds. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; 2001:3421-52.
- Ohto U, Usui K, Ochi T, Yuki K, Satow Y, Shimizu T.Crystal structure of human β-galactosidase: structural basis of Gm1 gangliosidosis and Morquio B diseases. J Biol Chem. 2012; 287:1801-12. [PMC free article: PMC3265862] [PubMed: 22128166]
- Onçağ G, Ertan Erdinç AM, Cal E. Multidisciplinary treatment approach of Morquio syndrome (mucopolysaccharidosis type IVA). Angle Orthod. 2006;76:335-40. [PubMed: 16539564]
- Peracha H, Sawamoto K, Averill L, Kecskemethy H, Theroux M, Thacker M, Nagao K, Pizarro C, Mackenzie W, Kobayashi H, Yamaguchi S, Suzuki Y, Orii K, Orii T, Fukao T, Tomatsu S. Molecular genetics and metabolism, special edition: Diagnosis, diagnosis and prognosis of mucopolysaccharidosis IVA. Mol Genet Metab. 2018;125:18-37. [PMC free article: PMC6175643] [PubMed: 29779902]
- Pruszczynski B, Mackenzie WG, Rogers K, White KK. Spinal cord injury after extremity surgery in children with thoracic kyphosis. Clin Orthop Relat Res. 2015;473:3315-20. [PMC free article: PMC4562919] [PubMed: 26242281]
- Ransford AO, Crockard HA, Stevens JM, Modaghegh S. Occipito-atlanto-axial fusion in Morquio-Brailsford syndrome. A ten-year experience. J Bone Joint Surg Br. 1996;78:307-13. [PubMed: 8666648]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rivera-Colón Y, Schutsky EK, Kita AZ, Garman SC. The structure of human GALNS reveals the molecular basis for mucopolysaccharidosis IV A. J Mol Biol. 2012;423:736-51. [PMC free article: PMC3472114] [PubMed: 22940367]
- Rodriguez ME, Mackenzie WG, Ditro C, Miller TL, Chidekel A, Shaffer TH. Skeletal dysplasias: evaluation with impulse oscillometry and thoracoabdominal motion analysis. Pediatr Pulmonol. 2010;45:679-86. [PMC free article: PMC3338356] [PubMed: 20575094]
- Ruckenstein MJ, Macdonald RE, Clarke JT, Forte V. The management of otolaryngological problems in the mucopolysaccharidoses: a retrospective review. J Otolaryngol. 1991;20:177-83. [PubMed: 1908026]
- Sawamoto K, Álvarez González JV, Piechnik M, Otero FJ, Couce ML, Suzuki Y, Tomatsu S. Mucopolysaccharidosis IVA: diagnosis, treatment, and management. Int J Mol Sci. 2020;21:1517. [PMC free article: PMC7073202] [PubMed: 32102177]
- Semenza GL, Pyeritz RE. Respiratory complications of mucopolysaccharide storage disorders. Medicine (Baltimore). 1988;67:209-19. [PubMed: 3134589]
- Simmons MA, Bruce IA, Penney S, Wraith E, Rothera MP. Otorhinolaryngological manifestations of the mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol. 2005;69:589-95. [PubMed: 15850680]
- Solanki GA, Martin KW, Theroux MC, Lampe C, White KK, Shediac R, Lampe CG, Beck M, Mackenzie WG, Hendriksz CJ, Harmatz PR. Spinal involvement in mucopolysaccharidosis IVA (Morquio-Brailsford or Morquio A syndrome):presentation, diagnosis and management. J. Inherit Metab Dis. 2013;36:339-55. [PMC free article: PMC3590412] [PubMed: 23385297]
- Theroux MC, Nerker T, Ditro C, Mackenzie WG. Anesthetic care and perioperative complications of children with Morquio syndrome. Paediatr Anaesth. 2012;22:901-7. [PubMed: 22738181]
- Tomatsu S, Montaño AM, Nishioka T, Gutierrez MA, Peña OM, Tranda Firescu GG, Lopez P, Yamaguchi S, Noguchi A, Orii T . Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A). Hum Mutat. 2005;26:500-12. [PubMed: 16287098]
- Tomatsu S, Montaño AM, Oikawa H, Smith M, Barrera L, Chinen Y, Thacker MM, Mackenzie WG, Suzuki Y, Orii T. Mucopolysaccharidosis type IVA (Morquio A disease): clinical review and current treatment. Curr Pharm Biotechnol. 2011;12:931-45. [PubMed: 21506915]
- Tong CK, Chen JC, Cochrane DD. Spinal cord infarction remote from maximal compression in a patient with Morquio syndrome. J Neurosurg Pediatr. 2012;9:608-12. [PubMed: 22656250]
- Walker PP, Rose E, Williams JG. Upper airways abnormalities and tracheal problems in Morquio's disease. Thorax. 2003;58:458-9. [PMC free article: PMC1746674] [PubMed: 12728175]
- Walker R, Belani KG, Braunlin EA, Bruce IA, Hack H, Harmatz PR, Jones S, Rowe R, Solanki GA, Valdemarsson B. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36:211-9. [PMC free article: PMC3590422] [PubMed: 23197104]
- White KK, Jester A, Bache CE, Harmatz PR, Shediac R, Thacker MM, Mackenzie WG.Orthopedic management of the extremities in patients with Morquio A syndrome. J Child Orthop. 2014;8:295-304. [PMC free article: PMC4128951] [PubMed: 25001525]
- Wilson W, Taubert KA, Gewitz M, Lockhart PB, Baddour LM, Levison M, Bolger A, Cabell CH, Takahashi M, Baltimore RS, Newburger JW, Strom BL, Tani LY, Gerber M, Bonow RO, Pallasch T, Shulman ST, Rowley AH, Burns JC, Ferrieri P, Gardner T, Goff D, Durack DT, et al. Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007;116:1736-54. [PubMed: 17446442]
- Wood TC, Harvey K, Beck M, Burin MG, Chien YH, Church HJ, D'Almeida V, van Diggelen OP, Fietz M, Giugliani R, Harmatz P, Hawley SM, Hwu WL, Ketteridge D, Lukacs Z, Miller N, Pasquali M, Schenone A, Thompson JN, Tylee K, Yu C, Hendriksz CJ. Diagnosing mucopolysaccharidosis IVA. J Inherit Metab Dis. 2013;36:293-307. [PMC free article: PMC3590423] [PubMed: 23371450]
- Wraith JE. The mucopolysaccharidoses: a clinical review and guide to management Arch Dis Child.1995;72:263-7. [PMC free article: PMC1511064] [PubMed: 7741581]
- Zanetti A, D'Avanzo F, Rigon L, Rampazzo A, Concolino D, Barone R, Volpi N, Santoro L, Lualdi S, Bertola F, Scarpa M, Tomanin R. Molecular diagnosis of patients affected by mucopolysaccharidosis: a multicenter study. Eur J Pediatr. 2019;178:739-53. [PMC free article: PMC6459791] [PubMed: 30809705]
Publication Details
Author Information and Affiliations
Washington, DC
Washington, DC
Washington, DC
Publication History
Initial Posting: July 11, 2013; Last Update: June 17, 2021.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Regier DS, Oetgen M, Tanpaiboon P. Mucopolysaccharidosis Type IVA. 2013 Jul 11 [Updated 2021 Jun 17]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.