Summary
Clinical characteristics.
Maple syrup urine disease (MSUD) is categorized as classic (severe), intermediate, or intermittent. Neonates with classic MSUD are born asymptomatic but without treatment follow a predictable course:
- 12–24 hours. Elevated concentrations of branched-chain amino acids (BCAAs; leucine, isoleucine, and valine) and alloisoleucine, as well as a generalized disturbance of amino acid concentration ratios, are present in blood and the maple syrup odor can be detected in cerumen;
- Two to three days. Early and nonspecific signs of metabolic intoxication (i.e., irritability, hypersomnolence, anorexia) are accompanied by the presence of branched-chain alpha-ketoacids, acetoacetate, and beta-hydroxybutyrate in urine;
- Four to six days. Worsening encephalopathy manifests as lethargy, apnea, opisthotonos, and reflexive "fencing" or "bicycling" movements as the sweet maple syrup odor becomes apparent in urine;
- Seven to ten days. Severe intoxication culminates in critical cerebral edema, coma, and central respiratory failure.
Individuals with intermediate MSUD have partial branched-chain alpha-ketoacid dehydrogenase deficiency that manifests only intermittently or responds to dietary thiamine therapy; these individuals can experience severe metabolic intoxication and encephalopathy in the face of sufficient catabolic stress. In the era of newborn screening (NBS), the prompt initiation of treatment of asymptomatic infants detected by NBS means that most individuals who would have developed neonatal manifestations of MSUD remain asymptomatic with continued treatment adherence.
Diagnosis/testing.
Suggestive biochemical findings on NBS include whole-blood concentration ratios of (leucine + isoleucine) to alanine and phenylalanine that are above the cutoff values for the particular screening lab. Follow-up plasma amino acid analysis typically demonstrates elevated concentrations of BCAAs and alloisoleucine. The diagnosis of MSUD is confirmed by identification of biallelic pathogenic variants in BCKDHA, BCKDHB, or DBT.
Management.
Treatment of manifestations: Treatment consists of dietary leucine restriction, BCAA-free medical foods, judicious supplementation with isoleucine and valine, and frequent clinical and biochemical monitoring. A BCAA-restricted diet fortified with prescription medical foods can maintain average plasma BCAA concentrations within standard reference intervals and preserves the ratios among them. Use of a "sick-day" formula recipe (devoid of leucine and enriched with calories, isoleucine, valine, and BCAA-free amino acids) combined with rapid and frequent amino acid monitoring allows many catabolic illnesses to be managed in the outpatient setting. Acute metabolic decompensation is corrected by treating the precipitating stress while delivering sufficient calories, insulin, free amino acids, isoleucine, and valine to achieve sustained net protein synthesis in tissues. Some centers use hemodialysis/hemofiltration to remove BCAAs from the extracellular compartment, but this intervention does not alone establish net protein accretion. Brain edema is a common complication of metabolic encephalopathy and requires careful management in an intensive care setting. Adolescents and adults with MSUD are at increased risk for attention-deficit/hyperactivity disorder, depression, and anxiety disorders and can be treated successfully with standard psychostimulant and antidepressant medications.
Prevention of primary manifestations: Transplantation of allogeneic liver tissue affords affected individuals an unrestricted diet and protects them from metabolic crises, but does not reverse preexisting psychomotor disability or mental illness. In those who have not undergone liver transplantation, strict and consistent metabolic control can decrease the risk of developing neuropsychiatric morbidities. Consider a trial of enteral thiamine to determine if an affected individual may have thiamine-responsive disease.
Prevention of secondary complications: Any trauma care or surgical procedures should be approached in consultation with a metabolic specialist.
Surveillance: Weekly or twice-weekly assessment of amino acid profile for rapidly growing infants; weekly amino acid profile assessment in children, adolescents, and adults; routine monitoring of calcium, magnesium, zinc, folate, selenium, and omega-3 essential fatty acid levels; at least monthly visit with a metabolic specialist in infancy; assessment of developmental milestones at each visit or as needed.
Evaluation of relatives at risk: It can be determined if newborn sibs of an affected individual (who have not been tested prenatally) are affected (1) by plasma amino acid analysis at approximately 24 hours of life; or (2) by molecular genetic testing of umbilical cord blood if the family-specific pathogenic variants have been identified. Early diagnosis may allow management of asymptomatic infants out of hospital by experienced providers. Before confirmatory molecular testing is complete, at-risk neonates can be managed with an MSUD prescription diet if serial plasma amino acid profiles provide evidence of MSUD.
Pregnancy management: For women with MSUD, metabolic control should be rigorously maintained before and throughout pregnancy by frequent monitoring of plasma amino acid concentrations and dietary adjustments to avoid the likely teratogenic effects of elevated maternal leucine plasma concentration. Fetal growth should be monitored to detect any signs of essential amino acid deficiency. The catabolic stress of labor, involutional changes of the uterus, and internal sequestration of blood are potential sources of metabolic decompensation of the affected mother. Appropriate monitoring of the affected mother at a metabolic referral center at the time of delivery and in the postpartum period are recommended.
Genetic counseling.
MSUD is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being unaffected and a carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk relatives and prenatal diagnosis for pregnancies at increased risk are possible if the pathogenic variants have been identified in an affected family member.
Diagnosis
Maple syrup urine disease (MSUD) is caused by decreased activity of the branched-chain alpha-ketoacid dehydrogenase complex (BCKD), the second enzymatic step in the degradative pathway of the branched-chain amino acids (BCAAs), which includes leucine, isoleucine, and valine.
Scenario 1. Abnormal newborn screening (NBS) result
- NBS for MSUD is primarily based on quantification of the ratios of (leucine + isoleucine) to alanine and phenylalanine concentrations on dry blood spots.
- A positive screening value (i.e., those above the cutoff reported by the screening laboratory) require follow-up biochemical testing with quantitative plasma amino acid and alloisoleucine analyses. If either is abnormal, treatment (see Management) and testing to establish the diagnosis (see Establishing the Diagnosis) should be initiated concurrently.Note: Individual states set standards for positive or suspected positive screens.
- Because leucine-isoleucine and hydroxyproline cannot be differentiated by mass spectrometry, neonates with isolated hydroxyprolinemia will screen positive for MSUD, but confirmatory amino acid analysis will show only increased hydroxyproline (a false positive newborn screening result).
- Neonates and infants suspected of having MSUD should never be challenged with higher than normal protein intake during the diagnostic process (see Management). This practice is dangerous; modern diagnostic methods make it unnecessary.
Scenario 2. A symptomatic individual with either atypical findings or untreated infantile-onset MSUD (resulting from any of the following: NBS not performed, false negative NBS result, or caregivers not adherent to recommended treatment following a positive NBS result)
Supportive clinical and laboratory findings can include the following.
Clinical findings
- Untreated infant:
- Maple syrup odor in cerumen, the first clinical sign of MSUD, is detectable 12 hours after birth.
- Signs of deepening encephalopathy including lethargy, intermittent apnea, opisthotonus, and stereotyped movements such as "fencing" and "bicycling" are evident by age four to five days.
- Coma and central respiratory failure may occur by age seven to ten days, sometimes before newborn screening results are available.
- Untreated older individuals with milder variants of MSUD:
- Anorexia
- Poor growth
- Irritability
- Developmental delays later in infancy or childhood
- Acute hyperleucinemia, ketonuria, and encephalopathy if stressed by fasting, dehydration, or infectious illness
Laboratory findings
- Elevated plasma concentrations of BCAAs and alloisoleucine
- Urinary excretion of BCKDs and branched-chain alpha-ketoacids (BCKAs) in infants older than 48-72 hours on an unrestricted diet
- Ketonuria detected by standard urine test stripsKetonuria in a newborn should always prompt investigation for metabolic disorders.
- Absence of hypoglycemia and hyperammonemia
Establishing the Diagnosis
The diagnosis of MSUD in a proband with suggestive metabolic/biochemical findings is established by identification of biallelic pathogenic (or likely pathogenic) variants in one of the genes listed in Table 1 or – in limited instances – by significantly reduced activity of the BCKD enzyme in cultured fibroblasts, leukocytes, or biopsied liver tissue. Because of its relatively high sensitivity, molecular genetic testing can obviate the need for enzymatic testing and, thus, is increasingly the preferred confirmatory test for MSUD.
Note: (1) In vitro measurements of BCKD activity do not correlate with measurements of in vivo leucine oxidation [Schadewaldt et al 2001], dietary leucine tolerance [Strauss et al 2010], or in vivo response to BCKD-activating medications [Brunetti-Pierri et al 2011]. Therefore, the authors do not find measurements of BCKD enzyme activity clinically useful. (2) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (3) Identification of a heterozygous variant of uncertain significance in one of the genes listed in Table 1 does not establish or rule out the diagnosis.
Molecular Genetic Testing Approaches
Scenario 1. Abnormal newborn screening (NBS) result. When NBS results and other laboratory findings suggest the diagnosis of MSUD, the preferred molecular genetic testing approach is use of a multigene panel that contains BCKDHA, BCKDHB, and DBT. In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
- Sequence analysis detects missense, nonsense, and splice site variants and small intragenic deletions/insertions. Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected.
- For this disorder, a multigene panel that also includes deletion/duplication analysis is recommended (see Table 1).
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Scenario 2. A symptomatic individual with atypical findings or untreated infantile-onset MSUD (resulting from NBS not performed or false negative NBS result). When the diagnosis of MSUD has not been considered, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible. If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Maple Syrup Urine Disease
Clinical Characteristics
Clinical Description
Traditionally, the metabolic phenotype of maple syrup urine disease (MSUD) is termed classic or intermediate on the basis of residual branched-chain alpha-ketoacid dehydrogenase (BCKD) enzyme activity. Rarely, affected individuals have partial BCKD enzyme deficiency that manifests only intermittently or responds to dietary thiamine therapy (see Table 2). Phenotypic distinctions are not absolute: individuals with intermediate or intermittent forms of MSUD can experience severe metabolic intoxication and encephalopathy if physiologic stress is sufficient to overwhelm residual BCKD activity or this activity is reduced by transient changes in the phosphorylation state of the enzyme complex. Even in persons with relatively high baseline residual BCKD enzyme activity, episodes of metabolic intoxication can be fatal.
Table 2.
Clinical Phenotypes of Maple Syrup Urine Disease
Metabolic considerations in establishing MSUD phenotype:
- Dietary leucine tolerance. Leucine tolerance is defined as the weight-adjusted daily leucine intake that is sufficient for normal growth and maintains plasma leucine concentration within the normal range (reference mean ±2 SDs). In persons with classic MSUD, in vivo oxidation and urinary losses of branched-chain amino acids (BCAAs) are negligible [Schadewaldt et al 1999b, Levy 2001]. Thus, leucine tolerance reflects a balance between unmeasured protein losses (e.g., sloughed skin, hair, and nails) and the net accretion of body protein, which in turn is linked to growth rate [Strauss et al 2010]. During metabolic crises, changes of plasma leucine mirror whole-body protein turnover, which can be quantified if one assumes the human body is 10%-12% protein, protein is about 10% leucine by weight, and free leucine (molecular weight 131 mg/mmol) is evenly distributed in total body water [Garrow et al 1965, Filho et al 1997].
- Plasma concentration ratios of BCAAs. The wild type BCKD complex maintains stringent stoichiometric relationships among the three BCAAs, such that plasma concentration ratios (µmol/L:µmol/L) of leucine to isoleucine (Leu/Iso) and valine to leucine (Val/Leu) remain close to 2.0 in diverse physiologic contexts, including overnight fasting, protein loading, and catabolic illness. In contrast, these concentration ratios vary across several orders of magnitude in individuals with classic MSUD [Strauss et al 2006]. Individuals with intermediate forms of MSUD are less vulnerable to these volatile changes of plasma BCAA concentrations and less likely to experience prolonged essential amino acid deficiencies.
- Plasma alloisoleucine. Alloisoleucine is a chemical derivative of isoleucine and represents the most sensitive and specific diagnostic marker for all forms of MSUD. Plasma alloisoleucine is <5 µmol/L in healthy infants, children, and adults, and exceeds this value in 94% and 99.9% of samples from individuals with intermediate and classic forms of MSUD, respectively [Schadewaldt et al 1999a].
- Rapidity and severity of decompensation during illness. The risk for metabolic crisis in any ill person with MSUD depends on residual in vivo BCKD enzyme activity in relation to the net liberation of free leucine from protein catabolism. Thus, individuals with residual in vivo BCKD enzyme activity enjoy a higher leucine tolerance when well and also tend to have slower and less severe elevations of plasma leucine concentrations during illnesses.
Classic MSUD Phenotype
Maple syrup odor is evident in cerumen soon after birth and in urine by age five to seven days. In untreated neonates, ketonuria, irritability, and poor feeding occur within 48 hours of delivery. Lethargy, intermittent apnea, opisthotonus, and stereotyped movements such as "fencing" and "bicycling" are evident by age four to five days and are followed by coma and central respiratory failure. Preemptive detection of affected newborns, before they exhibit neurologic signs of MSUD, significantly reduces lifetime risk of intellectual disability, mental illness, and global functional impairment [Strauss et al 2012, Muelly et al 2013, Strauss et al 2020].
Following the neonatal period, acute metabolic intoxication (leucinosis) and neurologic deterioration can develop rapidly at any age as a result of net protein degradation precipitated by infection, surgery, injury, or psychological stress (see Figure 1). In infants and toddlers, leucinosis causes nausea, anorexia, altered level of consciousness, acute dystonia, and ataxia. Neurologic signs of intoxication in older individuals vary and can include cognitive impairment, hyperactivity, sleep disturbances, hallucinations, mood swings, focal dystonia, choreoathetosis, and ataxia. As plasma concentrations of leucine and alpha-ketoisocaproic acid (aKIC) increase, individuals become increasingly stuporous and may progress to coma. In persons of all ages with MSUD, nausea and vomiting are common during crisis and often necessitate hospitalization [Morton et al 2002].
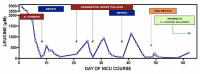
Each episode of acute leucinosis is associated with a risk for cerebral edema (see Figure 2) [Levin et al 1993] and death [Strauss et al 2020]. Mechanisms of brain edema in MSUD are not completely understood. Plasma leucine concentration correlates only indirectly with the degree of swelling; severe cerebral edema and neurologic impairment are more directly related to the rate of change of plasma leucine and concomitant decreases in blood osmolarity. During the evolution of leucinosis, cerebral vasopressin release may be provoked by both acute hyperosmolarity (from the accumulation of BCAAs, ketoacids, ketone bodies, and free fatty acids in the circulation) and vomiting. Renal excretion of branched-chain alpha-ketoacids (BCKAs) is accompanied by obligate urine sodium loss, and when this coincides with renal free water retention (antidiuresis), administration of hypotonic or even isotonic fluids can result in hyponatremia and critical brain edema [Strauss & Morton 2003].
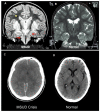
Transient periods of MSUD encephalopathy appear fully reversible, provided no global or focal ischemic brain damage occurs. In contrast, prolonged amino acid imbalances, particularly if they occur during the early years of brain development, lead to structural and functional neurologic abnormalities that have morbid long-term psychomotor consequences [Carecchio et al 2011, Shellmer et al 2011, Muelly et al 2013, Strauss et al 2020].
Neonatal screening and sophisticated enteral and parenteral treatment protocols (see Management) have significantly improved neurologic outcomes for persons with classic MSUD [Strauss et al 2010, Muelly et al 2013, Strauss et al 2020], but risks of acute brain injury or death are always present, and the long-term neuropsychiatric prognosis is guarded. In two longitudinal studies of individuals with classic MSUD [Muelly et al 2013, Strauss et al 2020], an asymptomatic neonatal course and stringent longitudinal biochemical control proved fundamental to optimizing long-term cognitive outcome and mental health.
- Early developmental milestones. Children with MSUD who are diagnosed during the neonatal period and managed prospectively under stringent dietary control can achieve major developmental milestones along time courses similar to their unaffected sibs [Strauss et al 2020].
- Cognitive function. Among individuals with classic MSUD (n = 81, ages 3.6-51.1 years), full scale intelligence quotient (FSIQ) correlates with birthdate (rs = 0.39, p = 0.0044) and is on average 20%-40% lower in affected individuals as compared to their unaffected sibs [Strauss et al 2020]. This difference is most striking for affected individuals born before the advent of newborn screening (NBS) (FSIQ of 62 ± 17, range 40-99). FSIQ correlates directly with the frequency of amino acid monitoring and inversely with both average lifetime plasma leucine and its concentration ratio to valine [Muelly et al 2013]. Prolonged neonatal encephalopathy is the single strongest predictor of neurocognitive disability and global functional impairment [Muelly et al 2013, Strauss et al 2020].
- Mood and anxiety. Among individuals with classic MSUD who complete appropriate objective testing, the probability of affective illness (depression, anxiety, and panic disorder) is between 83% and 100% by age 35 years [Muelly et al 2013, Strauss et al 2020]. Newborns who were encephalopathic at the time of diagnosis are five and ten times more likely, respectively, to later suffer from anxiety and depression (see Table 3) [Muelly et al 2013].
- Attention and hyperactivity. Cumulative lifetime incidence of attention-deficit/hyperactivity disorder (ADHD) exceeds 50% among individuals with MSUD on dietary therapy and may be even higher among those who underwent liver transplantation [Muelly et al 2013].
- Movement disorders. Among 17 adults with MSUD (mean age 27.5 years), 12 (70.6%) had a movement disorder (primarily tremor, dystonia, or a combination of both) on clinical examination [Carecchio et al 2011]. Parkinsonism and simple motor tics were also observed. Pyramidal signs were present in 11 affected individuals (64.7%), and a spastic-dystonic gait was observed in six (35.2%). In the authors' experience, such motor disabilities are rare in individuals with MSUD who are managed appropriately from the neonatal period but common among those who did not have the advantage of NBS [Strauss et al 2020].
Table 3.
Lifetime Relative Risk of Each Finding Based on Condition at the Time of Diagnosis
Liver transplantation appears to prevent catastrophic brain injuries that can occur during metabolic intoxication [Mazariegos et al 2012] and arrests the progression of neurocognitive impairment [Shellmer et al 2011], but does not reverse preexisting cognitive disability or psychiatric illness [Strauss et al 2020]. Neuropsychiatric morbidity and neurochemistry are similar among individuals with MSUD who have and have not undergone liver transplantation [Muelly et al 2013, Strauss et al 2020].
Non-central nervous system involvement in MSUD can include:
- Iatrogenic essential amino acid deficiency. Anemia, acrodermatitis, hair loss, growth failure, arrested head growth, anorexia, and lassitude are complications of chronic deficiency of leucine, isoleucine, or valine [Puzenat et al 2004]. Iatrogenic cerebral essential amino acid deficiency can be a cause of significant neurologic morbidity in any individual ingesting a diet low in natural protein and high in prescription medical protein [Strauss et al 2010, Manoli et al 2016].
- Recurrent oroesophageal candidiasis. Candida infections are common in hospitalized persons with MSUD and may result from T-cell inhibitory effects of elevated plasma leucine [Hidayat et al 2003] or iatrogenic immunodeficiency as a result of inadequate BCAA intake.
Intermediate MSUD
Similar principles govern the acute and chronic management of classic and intermediate forms of MSUD (see Management), and the distinction between them is not absolute (see Genotype-Phenotype Correlations) [Strauss et al 2020]. Individuals with residual BCKD activity (i.e., 3%-30% ex vivo) may appear well during the neonatal period but nevertheless have maple syrup odor in cerumen and a consistently abnormal plasma amino acid profile (see Table 2). Individuals with intermediate MSUD can present with feeding problems, poor growth, and developmental delay during infancy, or may present much later in life with apparently nonsyndromic intellectual disability [Chuang & Shih 2001]. The majority of persons with intermediate MSUD are detected by NBS, although detection later in childhood can occur in settings where newborns are not tested for MSUD. When followed longitudinally, individuals with intermediate forms of MSUD have plasma BCAA concentrations similar to those observed in individuals with the classic form, but tolerate more dietary leucine and require less nutritional support to reverse episodes of metabolic intoxication [Strauss et al 2020]. Children and adults with intermediate MSUD can nevertheless develop severe leucinosis and brain swelling if subjected to sufficient catabolic stress.
Intermittent MSUD
Children with the intermittent form of MSUD have normal growth and intellectual development throughout infancy and early childhood. When they are well, they generally tolerate a normal leucine intake, and plasma amino acid and urine organic acid profiles are normal or show only mild elevations of BCAAs. During infections or other physiologic stress, they can develop the clinical and biochemical features of classic MSUD, in rare cases culminating in coma and death [Chuang & Shih 2001]. These individuals may escape detection by NBS.
Thiamine-Responsive MSUD
It is not known with certainty if individuals with true thiamine-responsive MSUD exist. In general, such putative individuals have residual ex vivo BCKD enzyme activity of up to 40% normal and are not ill in the neonatal period, but present later in life with a clinical course similar to intermediate MSUD. To date, no person with "thiamine-responsive" MSUD has been treated solely with thiamine. Rather, they are treated with a combination of thiamine (doses ranging from 10 to 1,000 mg/day) and dietary BCAA restriction, making the in vivo contribution of thiamine impossible to discern [Chuang et al 2004]. Based on in vitro data, Chuang et al [2006] provided a biochemical model of thiamine responsiveness linked to specific pathogenic variants in the E2 subunit of BCKD. It is therefore reasonable to try thiamine supplementation under controlled dietary conditions in any individual with MSUD who has verified BCKDHB pathogenic variants.
Pathophysiology
BCKD has four subunit components (E1a, E1b, E2, and E3). Pathogenic variants in both alleles encoding any subunit can result in decreased activity of the enzyme complex and the accumulation of BCAAs and corresponding BCKAs in tissues and plasma [Nellis et al 2003, Chuang et al 2004] (see Nomenclature).
For more information on the pathophysiology of MSUD click here (pdf).
Genotype-Phenotype Correlations
The severity of the MSUD metabolic phenotype is determined by the amount of residual BCKD enzyme activity relative to dietary BCAA excess and the large demands for BCAA oxidation that accompany fasting, illness, or other catabolic stresses [Strauss et al 2010]. Although there are some established relationships between genotype and biochemical phenotype (i.e., classic vs intermediate), clinical and functional outcomes (e.g., FSIQ, psychiatric illness, executive dysfunction) cannot be predicted from genotype [Strauss et al 2020]. Individuals with the same MSUD genotype may vary considerably in their cerebral response to metabolic crisis – some being more vulnerable than others to the complications of metabolic encephalopathy, brain edema, and mental illness – and long-term outcomes are largely related to the timing and quality of metabolic control.
Nomenclature
Biochemical derangement caused by biallelic pathogenic variants in BCKDHA encoding BCKA decarboxylase (E1) alpha subunit is sometimes referred to as MSUD type 1A. Biochemical derangement caused by biallelic pathogenic variants in BCKDHB encoding BCKA decarboxylase (E1) beta subunit is sometimes referred to as MSUD type 1B, and biallelic pathogenic variants in DBT encoding dihydrolipoyl transacylase (E2) subunit are sometimes referred to as MSUD type 2. All three are clinically indistinguishable biochemically.
Note: Dihydrolipoamide dehydrogenase deficiency, caused by biallelic pathogenic variants in DLD encoding the E3 subunit (lipoamide dehydrogenase) of BCKD, is sometimes referred to as MSUD type 3, although the phenotype is easily distinguishable from MSUD (see Differential Diagnosis).
Prevalence
MSUD is rare in most populations, with incidence estimates of 1:185,000 live births [Chuang & Shih 2001, Nellis et al 2003].
As a result of a founder variant (c.1312T>A) in BCKDHA (E1a), certain Mennonite populations of Pennsylvania, Kentucky, New York, Indiana, Wisconsin, Michigan, Iowa, and Missouri have a carrier frequency for classic MSUD as high as one in ten and a disease incidence of approximately one in 380 live births [Puffenberger 2003] (see Molecular Genetics).
Genetically Related (Allelic) Disorders
No other phenotypes are known to be associated with germline pathogenic variants in BCKDHA, BCKDHB, or DBT.
Differential Diagnosis
Entities to exclude in the encephalopathic neonate include birth asphyxia, hypoglycemia, status epilepticus, kernicterus, meningitis, and encephalitis. The few inborn errors of metabolism that present with neonatal encephalopathy include the following:
- Hyperketosis syndromes (e.g., beta-ketothiolase deficiency [OMIM 203750])
- Urea cycle defects (See Urea Cycle Disorders Overview.)
- Glycine encephalopathy (nonketotic hyperglycinemia)
- Propionic acidemia or isolated methylmalonic acidemia (rarely)
Among these, MSUD is unique for the sweet odor of cerumen and a positive urine dinitrophenylhydrazine test. Laboratory testing that includes quantitative plasma amino acids, plasma or whole-blood alloisoleucine, serum acylcarnitines, urine organic acids, plasma ammonia concentration, and serum lactate concentration distinguishes among these possibilities. In particular, quantitative analysis of plasma amino acids is generally sufficient to diagnosis MSUD expeditiously.
4,5-dimethyl-3-hydroxy-2[5H]-furanone (sotolone), which is thought to be responsible for the characteristic odor of MSUD [Podebrad et al 1999], is also found in maple syrup, fenugreek, and lovage. Maternal ingestion of fenugreek during pregnancy has resulted in false suspicion of MSUD [Korman et al 2001]. Topical benzoin, commonly used in NICUs, also gives off a strong sweet odor.
Note: Pathogenic variants in DLD, the gene encoding the E3 subunit, are associated with dihydrolipoamide dehydrogenase deficiency, which produces a different phenotype. Affected infants have hypotonia, developmental delay, dystonia/chorea, and a Leigh-type encephalopathy. BCKD enzyme activity is 0%-25% of control activity. Moderate elevations of plasma concentration of BCAAs, lactic acidemia, and hyperalaninemia are observed. In most cases, the disorder is lethal in infants.
Management
Evaluations Following Initial Diagnosis
When maple syrup urine disease (MSUD) is suspected during the diagnostic evaluation (i.e., due to elevated concentration of leucine, isoleucine, valine, and/or alloisoleucine), metabolic treatment should be initiated immediately.
Development and evaluation of treatment plans, training and education of affected individuals and their families, and avoidance of side effects of dietary treatment (i.e., malnutrition, growth failure) require a multidisciplinary approach to care with oversight and expertise from a specialized metabolic center. Consensus nutritional guidelines have been published [Frazier et al 2014] (full text) and two peer-reviewed articles provide general guidelines about the comprehensive treatment and monitoring of MSUD: see Strauss et al [2010] (full text), Strauss et al [2020] (full text).
To establish the extent of disease and needs in an individual following initial diagnosis of MSUD, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis of Maple Syrup Urine Disease
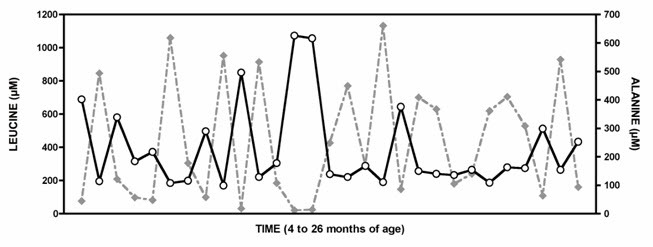
Figure 3.
Plasma amino values between ages four and 26 months from a child with classic MSUD show a strong reciprocal relationship between leucine (gray diamonds) and alanine (white circles) (Spearman correlation coefficient = -0.86; p<0.0001).
Republished with permission from Strauss et al [2010]
Treatment of Manifestations
All children with MSUD and feeding difficulties require supervision of a specialist metabolic dietitian with experience in managing the diet of those with MSUD.
The main principles of treatment are age-appropriate tolerance of leucine, isoleucine, and valine, with stable plasma branched-chain amino acid (BCAA) concentrations and BCAA concentration ratios, while avoiding deficiencies of essential amino acids, fatty acids, and micronutrients (see Table 5).
Table 5.
Routine Daily Treatment in Individuals with Maple Syrup Urine Disease
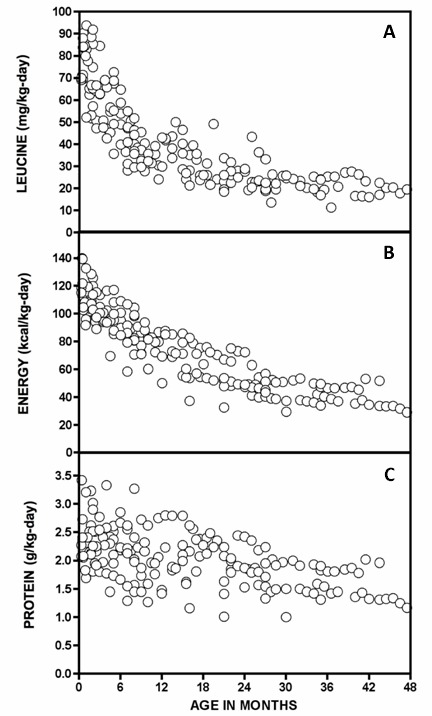
Figure 4.
Leucine (A), energy (B), and total protein (C) intakes of 15 stable Mennonite infants with classic MSUD on dietary management
Republished with permission from Strauss et al [2010]
Table 6.
Mean and 25th to 75th Percentile Range Nutrient Intakes (per kg-day) by Age Group
Mild illness. If an affected individual is clinically well despite an intercurrent infectious illness or febrile reaction to vaccinations, emergency outpatient management may be considered (see Table 7). If emergency outpatient treatment can be performed adequately and safely and if the child does not develop concerning symptoms during the illness, maintenance treatment and diet should be reintroduced stepwise over the next 48 (to 72) hours (see Table 5).
Table 7.
Emergency Outpatient Treatment in Individuals with Maple Syrup Urine Disease
Acute decompensation. Dietary indiscretion causes plasma BCAAs to increase but only rarely results in acute decompensation and encephalopathy. In contrast, infections and injuries trigger a large endogenous mobilization of muscle protein and can precipitate metabolic crisis and hospitalization.
Acute manifestations (e.g., lethargy, encephalopathy, seizures, or progressive coma) should be managed symptomatically and with generous caloric support in a hospital setting (see Table 8). Correction of metabolic decompensation is predicated on treating the underlying cause of the decompensation and establishing net protein accretion.
Table 8.
Acute Inpatient Treatment in Individuals with Maple Syrup Urine Disease
Prevention of Primary Manifestations
Table 9.
Prevention of Primary Manifestations in Individuals with Maple Syrup Urine Disease
Prevention of Secondary Complications
Any trauma care or surgical procedures should be approached in consultation with a metabolic specialist.
Surveillance
Table 10.
Recommended Surveillance for Individuals with Maple Syrup Urine Disease
Goals of laboratory monitoring
- Plasma leucine concentration: 150-300 µmol/L with an age-appropriate intake
- Plasma isoleucine concentration approximately equal to plasma leucine concentration
- Plasma valine concentration at least twofold plasma leucine concentration
- Indices of calcium, magnesium, zinc, folate, selenium, and omega-3 essential fatty acid sufficiency
Evaluation of Relatives at Risk
Early diagnosis of at-risk sibs of an affected individual may allow asymptomatic infants to be managed out of the hospital by experienced providers.
Newborn at-risk sibs who have not undergone prenatal testing can be tested in one of two ways:
- Plasma amino acid analysis of a sample obtained at approximately 24 hours of life. In some laboratories, samples obtained earlier can yield false negative results.
- If the pathogenic variants have been identified in the family, a cord blood sample can be used for molecular genetic testing.
Before confirmatory molecular testing is complete, at-risk neonates can be managed with an MSUD prescription diet if serial plasma amino acid profiles provide evidence of MSUD.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
With the advent of newborn screening and preventive care, more women with MSUD are surviving to child-bearing age. Successful delivery of a healthy baby is possible for women with classic MSUD [Van Calcar et al 1992, Grünewald et al 1998]. Two women who had biallelic pathogenic BCKDHA variants became pregnant following liver transplantation. Each delivered a healthy baby after remaining on immunosuppressant medication (sirolimus) but observing no special dietary restrictions during pregnancy [Strauss et al 2020].
Elevated maternal leucine plasma concentration, like elevated maternal phenylalanine plasma concentration, is likely teratogenic. If a woman with MSUD is planning a pregnancy, metabolic control should be maintained in a rigorous fashion preceding and throughout gestation. Keeping the maternal plasma levels of the branched-chain amino acids between 100 and 300 μmol/L is compatible with delivery of a normal infant [Grünewald et al 1998].
During the development of the placenta and fetus, maternal BCAA and protein requirements increase, and frequent monitoring of plasma amino acid concentrations and fetal growth may be necessary to avoid essential amino acid deficiencies [Grünewald et al 1998].
The postpartum period is dangerous for the affected mother. Catabolic stress of labor, involutional changes of the uterus, and internal sequestration of blood are potential sources of metabolic decompensation [Chuang & Shih 2001]. Appropriate monitoring at a metabolic referral center is advised at the time of delivery.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Maple syrup urine disease (MSUD) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., carriers of one BCKDHA, BCKDHB, or DBT pathogenic variant).
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. The offspring of an individual with maple syrup urine disease are obligate heterozygotes (carriers) for a BCKDHA, BCKDHB, or DBT pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a BCKDHA, BCKDHB, or DBT pathogenic variant.
Carrier Detection
Molecular genetic testing. Carrier testing for at-risk relatives requires prior identification of the BCKDHA, BCKDHB, or DBT pathogenic variants in the family.
Biochemical testing. Quantitative plasma amino acids and fibroblast enzymatic analyses are not indicated for detection of heterozygotes.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the BCKDHA, BCKDHB, or DBT pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing for a pregnancy at increased risk for MSUD are possible.
Biochemical testing. If only one or neither pathogenic variant is known within a family, branched-chain alpha-ketoacid dehydrogenase (BCKD) enzyme activity can be measured from cultured amniocytes obtained by amniocentesis (usually performed at ~15-18 weeks' gestation) or chorionic villus cells obtained by chorionic villus sampling (usually performed at ~10-12 weeks' gestation).
Note: Gestational age is expressed as menstrual weeks calculated either from the first day of the last normal menstrual period or by ultrasound measurements.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While use of prenatal testing is a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- British Inherited Metabolic Disease Group (BIMDG)TEMPLE (Tools Enabling Metabolic Parents LEarning)United Kingdom
- Medical Home Portal
- MSUD Family Support GroupThe MSUD Family Support Group is a non-profit 501 (c)(3) organization for those with MSUD and their families and includes health-care professionals and others interested in MSUD.Phone: 740-972-5619Email: sandybulcher@gmail.com
- National Library of Medicine Genetics Home Reference
- NCBI Genes and Disease
- Metabolic Support UKUnited KingdomPhone: 0845 241 2173
- Newborn Screening in Your StateHealth Resources & Services Administration
- Organic Acidemia AssociationPhone: 763-559-1797Email: info@oaanews.org
- European Registry and Network for Intoxication Type Metabolic Diseases (E-IMD)
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Maple Syrup Urine Disease: Genes and Databases
Table B.
OMIM Entries for Maple Syrup Urine Disease (View All in OMIM)
Molecular Pathogenesis
Maple syrup urine disease (MSUD) is caused by decreased activity of human branched-chain alpha-ketoacid dehydrogenase complex (BCKD), a multi-enzyme complex found in the mitochondria. It catalyzes the oxidative decarboxylation of the branched-chain ketoacids (alpha-ketoisocaproate, alpha-keto-beta-methyl valerate, and alpha-ketoisovalerate) in the second step in the degradative pathway of the branched-chain amino acids (BCAAs; leucine, isoleucine, and valine).
BCKD has four subunit components (E1a, E1b, E2, and E3). Biallelic pathogenic variants in one of the four unlinked genes encoding any subunit can result in decreased activity of the enzyme complex and the accumulation of BCAAs and corresponding branched-chain alpha-ketoacids (BCKAs) in tissues and plasma [Nellis et al 2003, Chuang et al 2004].
Biochemical derangements caused by pathogenic variants in the genes encoding BCKA decarboxylase (E1) alpha subunit (MSUD type 1A), BCKA decarboxylase (E1) beta subunit (MSUD type 1B), and dihydrolipoyl transacylase (E2) subunit (MSUD type 2) are indistinguishable biochemically.
Note: The E3 subunit (lipoamide dehydrogenase encoded by DLD) of BCKD is shared with the pyruvate and alpha-ketoglutarate dehydrogenase complexes, and MSUD type 3 is characterized by increased urinary excretion of BCKAs and alpha-ketoglutarate accompanied by elevated plasma concentrations of lactate, pyruvate, and alanine (see Table 2). However, the clinical phenotype of E3 subunit deficiency differs considerably from the classic and intermediate forms of MSUD and is not discussed further in this GeneReview.
The BCKD complex consists of three catalytic components:
- The E1 decarboxylase, which is a heterotetramer of alpha and beta subunits (alpha2, beta2)
- The E2 transacylase, which is a homo-24-mer
- The E3 dehydrogenase, which is a homodimer
The complete functional BCKD complex contains a cubic E2 core surrounded by the following:
- Twelve E1 components
- Six E3 components
- A single kinase
BCKD colocalizes with BCAA transaminases in mitochondria of diverse tissues and is regulated by a kinase-phosphatase pair. In humans, skeletal muscle is the major site for both transamination and oxidation of BCAAs. The liver and kidney each mediate an estimated 10%-15% of whole-body BCAA transamination-oxidation [Suryawan et al 1998]. BCKD is expressed in the brain, where BCAA transamination-oxidation may contribute to cerebral glutamate and GABA production [Yudkoff et al 2005].
Mechanism of disease causation. MSUD is a recessive disorder caused by biallelic pathogenic variants in BCKDHA, BCKDHB, or DBT that result in decreased function or loss of function.
Table 11.
Maple Syrup Urine Disease: Gene-Specific Laboratory Considerations
Table 12.
Maple Syrup Urine Disease: Notable Pathogenic Variants by Gene
Chapter Notes
Author History
D Holmes Morton, MD; Clinic for Special Children (2006-2020)
Erik G Puffenberger, PhD (2006-present)
Kevin A Strauss, MD (2006-present)
Vincent J Carson, MD (2020-present)
Revision History
- 23 April 2020 (ma) Comprehensive update posted live
- 9 May 2013 (me) Comprehensive update posted live
- 15 December 2009 (me) Comprehensive update posted live
- 30 January 2006 (me) Review posted live
- 22 November 2004 (ep) Original submission
References
Published Guidelines / Consensus Statements
- Frazier DM, Allgeier C, Homer C, Marriage BJ, Ogata B, Rohr F, Splett PL, Stembridge A, Singh RH. Nutrition management guideline for maple syrup urine disease: an evidence- and consensus-based approach. Available online. 2014. Accessed 5-25-22.
- Strauss KA, Carson VJ, Soltys K, Young ME, Bowser LE, Puffenberger EG, Brigatti KW, Williams KB, Robinson DL, Hendrickson C, Beiler K, Taylor CM, Haas-Givler B, Chopko S, Hailey J, Muelly ER, Shellmer DA, Radcliff Z, Rodrigues A, Loeven K, Heaps AD, Mazariegos GV, Morton DH. Branched-chain α-ketoacid dehydrogenase deficiency (maple syrup urine disease): Treatment, biomarkers, and outcomes. Available online. 2020. Accessed 5-25-22.
- Strauss KA, Wardley B, Robinson D, Hendrickson C, Rider NL, Puffenberger EG, Shellmer D, Moser AB, Morton DH. Classical maple syrup urine disease and brain development: principles of management and formula design. Mol Genet Metab. 99:333-45. Available online. 2010. Accessed 5-25-22.
Literature Cited
- Ali EZ, Ngu LH. Fourteen new mutations of BCKDHA, BCKDHB and DBT genes associated with maple syrup urine disease (MSUD) in Malaysian population. Mol Genet Metab Rep. 2018;17:22-30. [PMC free article: PMC6140420] [PubMed: 30228974]
- Bodner-Leidecker A, Wendel U, Saudubray JM, Schadewaldt P. (2000). Branched-chain L-amino acid metabolism in classical maple syrup urine disease after orthotopic liver transplantation. J Inherit Metab Dis. 23:805-18. [PubMed: 11196106]
- Boros Á, Pankovics P, Kőmíves S, Liptai Z, Dobner S, Ujhelyi E, Várallyay G, Zsidegh P, Bolba N, Reuter G. Co-infection with coxsackievirus A5 and norovirus GII.4 could have been the trigger of the first episode of severe acute encephalopathy in a six-year-old child with the intermittent form of maple syrup urine disease (MSUD). Arch Virol. 2017;162:1757-63. [PubMed: 28243803]
- Brodtkorb E, Strand J, Backe PH, Lund AM, Bjørås M, Rootwelt T, Rootwelt H, Woldseth B, Eide L. Four novel mutations identified in Norwegian patients result in intermittent maple syrup urine disease when combined with the R301C mutation. Mol Genet Metab. 2010;100:324-32. [PubMed: 20570198]
- Brunetti-Pierri N, Lanpher B, Erez A, Ananieva EA, Islam M, Marini JC, Sun Q, Yu C, Hegde M, Li J, Wynn RM, Chuang DT, Hutson S, Lee B. (2011). Phenylbutyrate therapy for maple syrup urine disease. Hum Mol Genet. 20:631-40. [PMC free article: PMC3024040] [PubMed: 21098507]
- Carecchio M, Schneider SA, Chan H, Lachmann R, Lee PJ, Murphy E, Bhatia KP. Movement disorders in adult surviving patients with maple syrup urine disease. Mov Disord. 2011;26:1324-8. [PMC free article: PMC4235248] [PubMed: 21484869]
- Chuang DT, Chuang JL, Wynn RM. (2006). Lessons from genetic disorders of branched-chain amino acid metabolism. J Nutr. 136:243S-9S. [PubMed: 16365091]
- Chuang DT, Fisher CW, Lau KS, Griffin TA, Wynn RM, Cox RP. Maple syrup urine disease: domain structure, mutations and exon skipping in the dihydrolipoyl transacylase (E2) component of the branched-chain alpha-keto acid dehydrogenase complex. Mol Biol Med. 1991;8:49-63. [PubMed: 1943690]
- Chuang DT, Shih VE. Maple syrup urine disease (branched-chain ketoaciduria). In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001:1971-2006.
- Chuang JL, Wynn RM, Moss CC, Song JL, Li J, Awad N, Mandel H, Chuang DT. (2004). Structural and biochemical basis for novel mutations in homozygous Israeli maple syrup urine disease patients: a proposed mechanism for the thiamin-responsive phenotype. J Biol Chem. 279:17792-800. [PubMed: 14742428]
- Edelmann L, Wasserstein MP, Kornreich R, Sansaricq C, Snyderman SE, Diaz GA. Maple syrup urine disease: identification and carrier-frequency determination of a novel founder mutation in the Ashkenazi Jewish population. Am J Hum Genet. 2001;69:863-8. [PMC free article: PMC1226071] [PubMed: 11509994]
- Fernández-Guerra P, Navarrete R, Weisiger K, Desviat LR, Packman S, Ugarte M, Rodríguez-Pombo P. Functional characterization of the novel intronic nucleotide change c.288+9C>T within the BCKDHA gene: understanding a variant presentation of maple syrup urine disease. J Inherit Metab Dis. 2010;33 Suppl 3:S191-8. [PubMed: 20431954]
- Filho JC, Bergström J, Stehle P, Fürst P. Simultaneous measurements of free amino acid patterns of plasma, muscle and erythrocytes in healthy human subjects. Clin Nutr. 1997;16:299-305. [PubMed: 16844612]
- Fisher CR, Fisher CW, Chuang DT, Cox RP. Occurrence of a Tyr393----Asn (Y393N) mutation in the E1 alpha gene of the branched-chain alpha-keto acid dehydrogenase complex in maple syrup urine disease patients from a Mennonite population. Am J Hum Genet. 1991;49:429-34. [PMC free article: PMC1683290] [PubMed: 1867199]
- Flaschker N, Feyen O, Fend S, Simon E, Schadewaldt P, Wendel U. Description of the mutations in 15 subjects with variant forms of maple syrup urine disease. J Inherit Metab Dis. 2007;30:903-9. [PubMed: 17922217]
- Frazier DM, Allgeier C, Homer C, Marriage BJ, Ogata B, Rohr F, Splett PL, Stembridge A, Singh RH. Nutrition management guideline for maple syrup urine disease: an evidence- and consensus-based approach. Mol Genet Metab. 2014;112:210-7. [PubMed: 24881969]
- Garrow JS, Fletcher K, Halliday D. Body composition in severe infantile malnutrition. J Clin Invest. 1965;44:417-25. [PMC free article: PMC292492] [PubMed: 14271301]
- Grünewald S, Hinrichs F, Wendel U. (1998). Pregnancy in a woman with maple syrup urine disease. J Inherit Metab Dis. 21:89-94. [PubMed: 9584259]
- Guo Y, Liming L, Jiang L. Two novel compound heterozygous mutations in the BCKDHB gene that cause the intermittent form of maple syrup urine disease. Metab Brain Dis. 2015;30:1395-400. [PubMed: 26239723]
- Henneke M, Flaschker N, Helbling C, Müller M, Schadewaldt P, Gärtner J, Wendel U. Identification of twelve novel mutations in patients with classic and variant forms of maple syrup urine disease. Hum Mutat. 2003;22:417. [PubMed: 14517957]
- Herring WJ, McKean M, Dracopoli N, Danner DJ. Branched chain acyltransferase absence due to an Alu-based genomic deletion allele and an exon skipping allele in a compound heterozygote proband expressing maple syrup urine disease. Biochim Biophys Acta. 1992;1138:236-42. [PubMed: 1547285]
- Hidayat S, Yoshino K, Tokunaga C, Hara K, Matsuo M, Yonezawa K. (2003). Inhibition of amino acid-mTOR signaling by a leucine derivative induces G1 arrest in Jurkat cells. Biochem Biophys Res Commun. 301:417-23. [PubMed: 12565877]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389-97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Jouvet P, Jugie M, Rabier D, Desgres J, Hubert P, Saudubray JM, Man NK. (2001). Combined nutritional support and continuous extracorporeal removal therapy in the severe acute phase of maple syrup urine disease. Intensive Care Med. 27:1798-806. [PubMed: 11810125]
- Jouvet P, Poggi F, Rabier D, Michel JL, Hubert P, Sposito M, Saudubray JM, Man NK. (1997). Continuous venovenous haemodiafiltration in the acute phase of neonatal maple syrup urine disease. J Inherit Metab Dis. 20:463-72. [PubMed: 9266382]
- Kahler SG, Sherwood WG, Woolf D, Lawless ST, Zaritsky A, Bonham J, Taylor CJ, Clarke JT, Durie P, Leonard JV. (1994). Pancreatitis in patients with organic acidemias. J Pediatr. 124:239-43. [PubMed: 8301430]
- Korman SH, Cohen E, Preminger A. (2001). Pseudo-maple syrup urine disease due to maternal prenatal ingestion of fenugreek. J Paediatr Child Health. 37:403-4. [PubMed: 11532065]
- Levin ML, Scheimann A, Lewis RA, Beaudet AL. (1993). Cerebral edema in maple syrup urine disease. J Pediatr. 122:167-8. [PubMed: 8419609]
- Levy HL. Hartnup disorder. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001:4957-70.
- Manoli I, Myles JG, Sloan JL, Carrillo-Carrasco N, Morava E, Strauss KA, Morton H, Venditti CP. (2016). A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 2: cobalamin C deficiency. Genet Med. 18:396-404. [PMC free article: PMC4752912] [PubMed: 26270766]
- Mazariegos GV, Morton DH, Sindhi R, Soltys K, Nayyar N, Bond G, Shellmer D, Shneider B, Vockley J, Strauss KA. Liver transplantation for classical maple syrup urine disease: long-term follow-up in 37 patients and comparative United Network for Organ Sharing experience. J Pediatr. 2012;160:116-21.e1. [PMC free article: PMC3671923] [PubMed: 21839471]
- Morton DH, Strauss KA, Robinson DL, Puffenberger EG, Kelley RI. (2002). Diagnosis and treatment of maple syrup disease: a study of 36 patients. Pediatrics. 109:999-1008. [PubMed: 12042535]
- Muelly ER, Moore GJ, Bunce SC, Mack J, Bigler DC, Morton DH, Strauss KA. (2013). Biochemical correlates of neuropsychiatric illness in maple syrup urine disease. J Clin Invest. 123:1809-20. [PMC free article: PMC3613932] [PubMed: 23478409]
- Nellis MM, Kasinski A, Carlson M, Allen R, Schaefer AM, Schwartz EM, Danner DJ. (2003). Relationship of causative genetic mutations in maple syrup urine disease with their clinical expression. Mol Genet Metab. 80:189-95. [PubMed: 14567968]
- Novarino G, El-Fishawy P, Kayserili H, Meguid NA, Scott EM, Schroth J, Silhavy JL, Kara M, Khalil RO, Ben-Omran T, Ercan-Sencicek AG, Hashish AF, Sanders SJ, Gupta AR, Hashem HS, Matern D, Gabriel S, Sweetman L, Rahimi Y, Harris RA, State MW, Gleeson JG. (2012). Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science. 338:394-7. [PMC free article: PMC3704165] [PubMed: 22956686]
- Nyhan WL, Rice-Kelts M, Klein J, Barshop BA. (1998). Treatment of the acute crisis in maple syrup urine disease. Arch Pediatr Adolesc Med. 152:593-8. [PubMed: 9641714]
- Podebrad F, Heil M, Reichert S, Mosandl A, Sewell AC, Bohles H. (1999). 4,5-dimethyl-3-hydroxy-2[5H]-furanone (sotolone)--the odour of maple syrup urine disease. J Inherit Metab Dis. 22:107-14. [PubMed: 10234605]
- Puckett RL, Lorey F, Rinaldo P, Lipson MH, Matern D, Sowa ME, Levine S, Chang R, Wang RY, Abdenur JE. Maple syrup urine disease: further evidence that newborn screening may fail to identify variant forms. Mol Genet Metab. 2010;100:136-42. [PubMed: 20307994]
- Puffenberger EG. (2003). Genetic heritage of the Old Order Mennonites of southeastern Pennsylvania. Am J Med Genet C Semin Med Genet. 121C:18-31. [PubMed: 12888983]
- Puliyanda DP, Harmon WE, Peterschmitt MJ, Irons M, Somers MJ. (2002). Utility of hemodialysis in maple syrup urine disease. Pediatr Nephrol. 17:239-42. [PubMed: 11956873]
- Puzenat E, Durbise E, Fromentin C, Humbert P, Aubin F. Iatrogenic acrodermatitis enteropathica-like syndrome in leucinosis. Ann Dermatol Venereol. 2004;131:801-4. [PubMed: 15505548]
- Quental S, Macedo-Ribeiro S, Matos R, Vilarinho L, Martins E, Teles EL, Rodrigues E, Diogo L, Garcia P, Eusébio F, Gaspar A, Sequeira S, Furtado F, Lança I, Amorim A, Prata MJ. Molecular and structural analyses of maple syrup urine disease and identification of a founder mutation in a Portuguese Gypsy community. Mol Genet Metab. 2008;94:148-56. [PubMed: 18378174]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rodríguez-Pombo P, Navarrete R, Merinero B, Gómez-Puertas P, Ugarte M. Mutational spectrum of maple syrup urine disease in Spain. Hum Mutat. 2006;27:715. [PubMed: 16786533]
- Schadewaldt P, Bodner-Leidecker A, Hammen HW, Wendel U. Whole-body L-leucine oxidation in patients with variant form of maple syrup urine disease. Pediatr Res. 2001;49:627-35. [PubMed: 11328944]
- Schadewaldt P, Bodner-Leidecker A, Hammen HW, Wendel U (1999a) Significance of L-alloisoleucine in plasma for diagnosis of maple syrup urine disease. Clin Chem. 45:1734-40. [PubMed: 10508118]
- Schadewaldt P, Hammen HW, Ott AC, Wendel U (1999b) Renal clearance of branched-chain L-amino and 2-oxo acids in maple syrup urine disease. J Inherit Metab Dis. 22:706-22. [PubMed: 10472531]
- Schaefer F, Straube E, Oh J, Mehls O, Mayatepek E (1999) Dialysis in neonates with inborn errors of metabolism. Nephrol Dial Transplant. 14:910-8. [PubMed: 10328469]
- Shellmer DA, DeVito Dabbs A, Dew MA, Noll RB, Feldman H, Strauss KA, Morton DH, Vockley J, Mazariegos GV. Cognitive and adaptive functioning after liver transplantation for maple syrup urine disease: a case series. Pediatr Transplant. 2011;15:58-64. [PMC free article: PMC3050647] [PubMed: 20946191]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197-207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Strauss KA, Carson VJ, Soltys K, Young ME, Bowser LE, Puffenberger EG, Brigatti KW, Williams KB, Robinson DL, Hendrickson C, Beiler K, Taylor CM, Haas-Givler B, Chopko S, Hailey J, Muelly ER, Shellmer DA, Radcliff Z, Rodrigues A, Loeven K, Heaps AD, Mazariegos GV, Morton DH. Branched-chain α-ketoacid dehydrogenase deficiency (maple syrup urine disease): treatment, biomarkers, and outcomes. Mol Genet Metab. 2020;129:193-206. [PubMed: 31980395]
- Strauss KA, Mazariegos GV, Sindhi R, Squires R, Finegold DN, Vockley G, Robinson DL, Hendrickson C, Virji M, Cropcho L, Puffenberger EG, McGhee W, Seward LM, Morton DH. (2006). Elective liver transplantation for the treatment of classical maple syrup urine disease. Am J Transplant. 6:557-64. [PubMed: 16468966]
- Strauss KA, Morton DH. (2003). Branched-chain ketoacyl dehydrogenase deficiency: maple syrup disease. Curr Treat Options Neurol. 5:329-41. [PubMed: 12791200]
- Strauss KA, Puffenberger EG, Morton DH. (2012). One community's effort to control genetic disease. Am J Public Health. 102:1300-6. [PMC free article: PMC3477994] [PubMed: 22594747]
- Strauss KA, Wardley B, Robinson D, Hendrickson C, Rider NL, Puffenberger EG, Shellmer D, Moser AB, Morton DH. (2010). Classical maple syrup urine disease and brain development: principles of management and formula design. Mol Genet Metab. 99:333-45. [PMC free article: PMC3671925] [PubMed: 20061171]
- Suryawan A, Hawes JW, Harris RA, Shimomura Y, Jenkins AE, Hutson SM. (1998). A molecular model of human branched-chain amino acid metabolism. Am J Clin Nutr. 68:72-81. [PubMed: 9665099]
- Szuch E, Auriemma J. (2018). Recurrent encephalopathy during febrile illnesses in a 6-year-old boy. Glob Pediatr Health. 2018;5:2333794X18784203. [PMC free article: PMC6041993] [PubMed: 30014008]
- Van Calcar SC, Harding CO, Davidson SR, Barness LA, Wolff JA. (1992). Case reports of successful pregnancy in women with maple syrup urine disease and propionic acidemia. Am J Med Genet. 44:641-6. [PubMed: 1481826]
- Wendel U, Saudubray JM, Bodner A, Schadewaldt P. (1999). Liver transplantation in maple syrup urine disease. Eur J Pediatr. 158 Suppl 2:S60-4. [PubMed: 10603101]
- Yoshino M, Aoki K, Akeda H, Hashimoto K, Ikeda T, Inoue F, Ito M, Kawamura M, Kohno Y, Koga Y, Kuroda Y, Maesaka H, Murakamisoda H, Sugiyama N, Suzuki Y, Yano S, Yoshioka A. (1999). Management of acute metabolic decompensation in maple syrup urine disease: a multi-center study. Pediatr Int. 41:132-7. [PubMed: 10221014]
- Yudkoff M, Daikhin Y, Nissim I, Horyn O, Luhovyy B, Lazarow A, Nissim I. Brain amino acid requirements and toxicity: the example of leucine. J Nutr. 2005;135:1531S-8S. [PubMed: 15930465]
- Zinnanti WJ, Lazovic J, Griffin K, Skvorak KJ, Paul HS, Homanics GE, Bewley MC, Cheng KC, Lanoue KF, Flanagan JM. (2009). Dual mechanism of brain injury and novel treatment strategy in maple syrup urine disease. Brain. 132:903-18. [PMC free article: PMC2668944] [PubMed: 19293241]
Publication Details
Author Information and Affiliations
Strasburg, Pennsylvania
Strasburg, Pennsylvania
Strasburg, Pennsylvania
Publication History
Initial Posting: January 30, 2006; Last Update: April 23, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Strauss KA, Puffenberger EG, Carson VJ. Maple Syrup Urine Disease. 2006 Jan 30 [Updated 2020 Apr 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

