Summary
The purpose of this overview is to increase the awareness of clinicians regarding the clinical characteristics of primary mitochondrial disorders, their diagnosis, and management, and to inform genetic counseling of individuals with a primary mitochondrial disorder and their family members. The following are the goals of this overview.
Goal 2.
Provide evaluation strategies to identify the genetic cause of a primary mitochondrial disorder in a proband.
Goal 4.
Inform genetic counseling of family members of an individual with a primary mitochondrial disorder.
1. Clinical Characteristics of Mitochondrial Disorders
Introduction
Primary mitochondrial disorders are a clinically heterogeneous group of disorders that arise as a result of dysfunction of the mitochondrial respiratory chain. The mitochondrial respiratory chain is the essential final common pathway for aerobic metabolism; tissues and organs that are highly dependent on aerobic metabolism are preferentially involved in mitochondrial disorders [Wallace 1999]. Many genetic and non-genetic disorders involve mitochondrial mechanisms as a secondary feature. However, "primary mitochondrial disorders" are considered to be known or presumed genetic disorders caused by pathogenic variants in genes coding for the mitochondrial respiratory chain and related proteins.
More than 70 different polypeptides interact on the inner mitochondrial membrane to form the respiratory chain. Thirteen essential subunits are encoded by mitochondrial DNA (mtDNA) located within mitochondria, along with the ribosomal and transfer RNAs required for intra-mitochondrial protein synthesis. The remaining respiratory chain polypeptides, and proteins essential for the assembly of the respiratory chain, mitochondrial structure, and the maintenance and expression of mtDNA are encoded by the nuclear genome (nDNA).
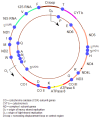
Mitochondrial DNA (mtDNA)
illustrates the structure of the human mitochondrial genome.
The human mitochondrial genome
The 1.1-kb D-loop (noncoding region) is involved in the regulation of transcription and replication of the molecule, and is the only region not directly involved in the synthesis of respiratory chain polypeptides.
The protein-encoding regions:
ND1 through ND6 and ND4L encode seven subunits of respiratory chain complex I.
CYT b encodes the only mtDNA-encoded respiratory chain complex III subunit.
CO I to CO III encode for three of respiratory chain complex IV (cytochrome c oxidase, or COX) subunits.
ATPase6 and ATPase8 encode for two subunits of respiratory chain complex V: ATPase6 and ATPase8, respectively.
Two ribosomal RNA genes (encoding 12S and 16S rRNA) and 22 transfer RNA genes are interspaced between the protein-encoding genes. These provide the necessary RNA components for intra-mitochondrial protein synthesis.
OH and OL are the origins of heavy- and light-strand mtDNA replication.
Each human cell contains thousands of copies of mtDNA. At birth these are usually all identical (homoplasmy). By contrast, individuals with mitochondrial disorders resulting from mtDNA pathogenic variants may harbor a mixture of mutated and wild type mtDNA within each cell (heteroplasmy) [Holt et al 1988, Holt et al 1990].
Single-cell studies have shown that the proportion of mtDNA pathogenic variants must exceed a critical threshold level before a cell expresses a biochemical abnormality of the mitochondrial respiratory chain (the threshold effect) [Schon et al 1997].
The percentage level of mtDNA pathogenic variants may vary among individuals within the same family, and also among organs and tissues within an individual [Macmillan et al 1993]. This is one explanation for the varied clinical phenotype seen in individuals with disorders caused by mtDNA pathogenic variants. For example, in individuals harboring the m.8993T>G pathogenic variant, higher percentage levels of this variant are seen in those with Leigh syndrome than in those with NARP (neurogenic weakness with ataxia and retinitis pigmentosa) [Uziel et al 1997, White et al 1999]. See Mitochondrial DNA-Associated Leigh Syndrome and NARP.
Nuclear DNA (nDNA)
Important mitochondrial mechanisms controlled by nuclear genes include the following:
Mitochondrial protein synthesis
Respiratory chain complexes
Respiratory chain assembly factors
Mitochondrial structure
Clinical Manifestations of Mitochondrial Disorders
Some mitochondrial disorders affect a single organ (e.g., the eye in Leber hereditary optic neuropathy and the ear in nonsyndromic hearing loss with or without aminoglycoside sensitivity; see Mitochondrial Nonsyndromic Hearing Loss and Deafness), whereas many involve multiple organ systems and often present with prominent neurologic and myopathic features.
Mitochondrial disorders may present at any age [Leonard & Schapira 2000a, Leonard & Schapira 2000b]. While initially it was generally thought that nDNA mitochondrial disorders presented in childhood and mtDNA disorders presented in late childhood or adulthood, recent advances have shown that many mtDNA disorders present in childhood, and many nDNA mitochondrial disorders present in adulthood.
Common clinical features of mitochondrial disorders include ptosis, external ophthalmoplegia, proximal myopathy and exercise intolerance, cardiomyopathy, sensorineural deafness, optic atrophy, pigmentary retinopathy, and diabetes mellitus. Diabetes mellitus and deafness is also a well-recognized clinical phenotype.
The central nervous system findings are often fluctuating encephalopathy, seizures, dementia, migraine, stroke-like episodes, ataxia, and spasticity. Chorea and dementia may also be prominent features.
A high incidence of mid- and late-pregnancy loss is also a common feature.
Mitochondrial DNA Disorders
Many individuals with a mtDNA disorder display a cluster of clinical features that fall into a discrete clinical syndrome (Table 1).
Table 1.
Clinical Syndromes of mtDNA Disorders
View in own window
| Disorder | Primary Features | Additional Features |
|---|
|
CPEO
| External ophthalmoplegia Bilateral ptosis
|
|
|
KSS
|
| Bilateral deafness Myopathy Dysphagia Diabetes mellitus Hypoparathyroidism Dementia
|
|
Pearson syndrome
|
|
|
|
Leigh syndrome
|
|
|
|
NARP
|
|
|
|
MELAS
|
|
|
|
MERRF
| Myoclonus Seizures Cerebellar ataxia Myopathy
| Dementia Optic atrophy Bilateral deafness Peripheral neuropathy Spasticity Multiple lipomata
|
|
LHON
|
|
|
CPEO = chronic progressive external ophthalmoplegia; KSS = Kearns-Sayre syndrome; LHON = Leber hereditary optic neuropathy; MELAS = mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes; MERRF = myoclonic epilepsy with ragged-red fibers; NARP = neurogenic weakness with ataxia and retinitis pigmentosa
Nuclear DNA Mitochondrial Disorders
Nuclear DNA (nDNA) mitochondrial disorders often display considerable clinical variability and many affected individuals do not fit neatly into one particular category. For example, mutation of POLG, which has emerged as a major cause of nDNA mitochondrial disorders, illustrates this well, with an overlapping spectrum of disease phenotypes (see POLG-Related Disorders).
Nuclear DNA mitochondrial disorders can be classified by disease mechanism (see Table 2).
Table 2.
Classification of Representative nDNA Mitochondrial Disorders
View in own window
| Genetic Cause | Disorders |
|---|
nDNA
disorders
of the mt
respiratory
chain
|
nDNA genes encoding structural subunits 1
| Leukodystrophy w/complex II deficiency Cardiomyopathy & encephalopathy (complex I deficiency) Optic atrophy & ataxia (complex II deficiency) Hypokalemia & lactic acidosis (complex III deficiency)
|
|
nDNA genes encoding assembly factors 1
| Hepatopathy & ketoacidosis Cardiomyopathy & encephalopathy Leukodystrophy & renal tubulopathy Hypertrophic cardiomyopathy Encephalopathy, liver failure, renal tubulopathy (w/complex III deficiency) Encephalopathy (w/complex V deficiency)
|
|
nDNA genes encoding translation factors 1
| Lactic acidosis, developmental failure, & dysmorphism Myopathy & sideroblastic anemia Leukodystrophy & polymicrogyria
|
|
nDNA disorders assoc w/multiple mtDNA deletions or mtDNA depletion
| Autosomal progressive external ophthalmoplegia Alpers-Huttenlocher syndrome Ataxia neuropathy syndromes 2 Mitochondrial encephalomyopathy w/combined RC deficiency Reversible hepatopathy Myopathy w/cataract & combined RC deficiency
|
Disorders of the
mt membrane
|
|
mt = mitochondrial; RC = respiratory chain
- 1.
- 2.
Includes MEMSA (myoclonic epilepsy myopathy sensory ataxia), MIRAS (mitochondrial recessive ataxia syndrome), SANDO (sensory ataxia neuropathy, dysarthria, ophthalmoplegia), and SCAE (spinocerebellar ataxia with epilepsy)
Secondary mitochondrial dysfunction in human diseases. Mitochondrial dysfunction is also seen in a number of different genetic disorders, including ethylmalonic aciduria (caused by mutation of ETHE1) [Tiranti et al 2009], Friedreich ataxia (FXN) [Rötig et al 1997], hereditary spastic paraplegia 7 (SPG7) [Casari et al 1998], and Wilson disease (ATP7B) [Lutsenko & Cooper 1998], and is also seen as part of the aging process. Because the term "mitochondrial disorder" usually refers to primary disorders of mitochondrial metabolism affecting oxidative phosphorylation, disorders manifesting secondary mitochondrial dysfunction are not strictly mitochondrial diseases and will not be discussed further in this chapter.
2. Evaluation Strategies to Identify the Genetic Cause of a Mitochondrial Disorder in a Proband
With more than 1,000 nuclear genes encoding mitochondrial proteins, the molecular diagnosis of mitochondrial disorders can be challenging. Establishing a molecular genetic diagnosis has important implications for the recurrence risk counseling of individuals with mitochondrial disease [Lieber et al 2013, Nesbitt et al 2013].
Mitochondrial dysfunction should be considered in the differential diagnosis of any progressive multisystem disorder. A full evaluation for a mitochondrial disorder is often warranted in individuals with a complex neurologic picture or a single neurologic manifestation and other system involvement.
Findings that can suggest a mitochondrial disorder include clinical phenotype (physical examination including neurologic examination), mode of inheritance (family history), and extent of the phenotype (other investigations to establish the extent of the phenotype).
Molecular genetic testing is used to establish the diagnosis.
Physical Examination and Neurologic Evaluation
A comprehensive physical examination is essential to identify asymptomatic organ involvement that would allow a syndromic diagnosis, or to identify asymptomatic complications that require management. Mitochondrial disorders can affect most organ systems, but a particular emphasis on the neuromuscular system and cardiovascular system is important.
Family History
A three-generation family history should be taken, with attention to relatives with manifestations of mitochondrial disorders and documentation of relevant findings through direct examination or review of medical records, including results of molecular genetic testing.
Simplex case. If only one member of a family is known to be affected, possibilities to consider include:
Autosomal dominant inheritance associated with a de novo pathogenic variant or decreased penetrance of a pathogenic variant associated with an autosomal dominant disorder
A single occurrence of an autosomal recessive, X-linked, or maternally inherited disorder
An acquired (non-genetic) cause
Many individuals with a childhood-onset encephalomyopathy and most adults with PEO or KSS represent simplex cases (Table 1).
Multiple affected family members
Mitochondrial inheritance. A family history in which males and females are affected, affected females transmit the disease to all their children, and affected males do not transmit the disease to their children suggests mitochondrial inheritance (see
Table 1). The range of clinical features associated with mtDNA pathogenic variants is broad and the family history may include many oligosymptomatic family members (e.g., some with diabetes mellitus or mild sensorineural deafness as the only feature).
Autosomal recessive inheritance. A family history in which sibs only are affected (i.e., a single generation in the family) and/or in which the parents are consanguineous suggests autosomal recessive inheritance.
Autosomal dominant inheritance. A family history in which males and females in multiple generations are affected suggests autosomal dominant inheritance. (Note: A clear autosomal dominant pattern of inheritance may be seen in individuals with PEO; however, absence of a known family history does not preclude the diagnosis.)
X-linked inheritance. A family history in which affected individuals are male and are related to each other through females (i.e., no male-to-male transmission) suggests X-linked inheritance.
Other Investigations to Establish the Extent of the Phenotype
Clinical tests are used to support a diagnosis of mitochondrial disease [Kaufmann et al 2009].
When the clinical picture is nonspecific but highly suggestive of a mitochondrial disorder, the clinician should start with measurement of plasma or CSF lactic acid concentration, ketone bodies, plasma acylcarnitines, and urinary organic acids.
Plasma/CSF lactate/pyruvate
Measurement of plasma lactate concentration is indicated in individuals with features of a myopathy or CNS disease. Fasting blood lactate concentrations above 3 mm/L support a diagnosis of mitochondrial disease.
Measurement of CSF lactate concentration is indicated in individuals with suspected CNS disease. Fasting CSF lactate concentrations above 1.5 mm/L support a diagnosis of mitochondrial disease.
Note: Normal plasma or CSF lactic acid concentration does not exclude the presence of a mitochondrial disorder.
Neuroimaging is indicated in individuals with suspected CNS disease. CT may show basal ganglia calcification and/or diffuse atrophy. MRI may show focal atrophy of the cortex or cerebellum, or high signal change on T2-weighted images, particularly in the occipital cortex [Scaglia et al 2005]. There may also be evidence of a generalized leukoencephalopathy [Barragán-Campos et al 2005]. Cerebellar atrophy is a prominent feature in children [Scaglia et al 2005].
Neurophysiologic studies
Electroencephalography (EEG) is indicated in individuals with suspected encephalopathy or seizures. Encephalopathy may be associated with generalized slow wave activity on EEG. Generalized or focal spike and wave discharges may be seen in individuals with seizures.
Peripheral neurophysiologic studies are indicated in individuals with limb weakness, sensory symptoms, or areflexia. Electromyography (EMG) is often normal but may show myopathic features. Nerve conduction velocity (NCV) may be normal or may show a predominantly axonal sensorimotor polyneuropathy.
Magnetic resonance spectroscopy (MRS) and exercise testing (with measurement of blood concentration of lactate) may be used to detect evidence of abnormal mitochondrial function noninvasively.
Glucose. An elevated concentration of fasting blood glucose may indicate diabetes mellitus.
Cardiac. Both electrocardiography and echocardiography may indicate cardiac involvement (cardiomyopathy or atrioventricular conduction defects).
Molecular Genetic Testing
Traditionally, the diagnosis of mitochondrial disorders has been based on demonstrating mitochondrial dysfunction in a relevant tissue biopsy (e.g., a skeletal muscle or liver biopsy, or skin fibroblasts), with the particular pattern of biochemical abnormality being used to direct targeted molecular genetic testing of mtDNA, specific nuclear genes, or both.
However, the more widespread availability of molecular diagnostic techniques and the advent of exome and genome sequencing has changed the diagnostic approach.
One important caveat arises from the fact that many mtDNA pathogenic variants are heteroplasmic, and the proportion of mutated mtDNA in blood may be undetectable. This can be circumvented by analyzing mtDNA from another tissue – typically skeletal muscle or urinary epithelium – in which the level of heteroplasmy tends to be higher. Some common mtDNA pathogenic variants (e.g., large-scale deletions causing CPEO) may only be detected in skeletal muscle.
Approaches to molecular genetic testing of a proband to consider are use of a multigene panel (Option 1) and comprehensive genomic testing (e.g., sequencing the mitochondrial genome; exome sequencing or genome sequencing to identify a pathogenic variant in a nuclear gene) (Option 2).
Option 1
In contrast to genomic testing, multigene panel testing relies on the clinician developing a hypothesis about which specific gene or set of genes to test. Hypotheses may be based on (1) mode of inheritance, (2) distinguishing clinical features, and/or (3) other discriminating features.
In individuals with a specific clinical phenotype (e.g., MELAS, LHON, POLG-related disorders) it may be possible to reach a diagnosis with targeted analysis of specific mtDNA pathogenic variants or single-gene testing of a nuclear gene. When these molecular tests do not identify the cause of the phenotype, or when the phenotype does not correspond to common clinical syndromes, many tertiary centers will perform biochemical tests on a tissue biopsy (see above), which can guide further molecular investigations. However, other laboratories immediately proceed to multigene panel testing, exome sequencing, or genome sequencing.
A mitochondrial disorders multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive
genomic testing does not require the clinician to determine which gene is likely involved. Such testing includes exome sequencing, genome sequencing, and mitochondrial sequencing which can simultaneously analyze nuclear DNA and mtDNA. However, as noted above, some mtDNA pathogenic variants may not be detected in leukocyte DNA.
Note: (1) False negative rates vary by genomic region; therefore, genomic testing may not be as accurate as targeted single-gene testing or multigene molecular genetic testing panels. (2) Most laboratories confirm positive results using a second, well-established method. (3) Certain DNA variants – such as large deletions or duplications (>8-10 bp in length), triplet repeat expansions, and epigenetic alterations – may not be detectable through genomic testing [Biesecker & Green 2014].
Other (Non-Molecular Genetic) Testing
In many individuals in whom molecular genetic testing does not yield or confirm a diagnosis, further investigation of suspected mitochondrial disease can involve a range of different clinical tests, including muscle biopsy for respiratory chain function.
3. Patient Care Guidelines
Several centers worldwide have produced guidelines on the clinical management of mitochondrial disease, including the international Mitochondrial Medicine Society [Parikh et al 2017]. These focus on regular surveillance to detect complications and their management using standard approaches that are not usually specific to mitochondrial disorders. Regular cardiac evaluation and monitoring for diabetes are particularly important.
There are currently no treatments known to influence the disease course in mitochondrial disease. Idebenone has been licensed as a treatment for LHON in some countries.
4. Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Mitochondrial disorders may be caused by mutation of a mitochondrial DNA (mtDNA) gene or mutation of a nuclear gene (nDNA).
mtDNA pathogenic variants are transmitted by maternal inheritance (mitochondrial inheritance).
nDNA pathogenic variants may be inherited in an autosomal recessive, autosomal dominant, or X-linked manner.
Note: The predisposition to form multiple mtDNA deletions can be inherited in an autosomal dominant or an autosomal recessive manner.
Mitochondrial Inheritance – Risk to Family Members
Parents of a proband
Sibs of a proband
The risk to the sibs depends on the genetic status of the mother: if the mother has the mtDNA pathogenic variant, all sibs will inherit it. If the mother of the proband is heteroplasmic for the mtDNA pathogenic variant, the proportion of mutated mtDNA she transmits to offspring varies.
Clinical expression in sibs depends on the proportion of mutated mtDNA (mutational load), and the organs and tissues in which they are found (tissue distribution and threshold effect). Sibs often inherit different proportions of mutated mtDNA, and thus can have a wide range of clinical manifestations.
When a proband has a single mtDNA deletion, the current best estimate of the recurrence risk to sibs is 1/24 [
Chinnery et al 2004].
Offspring of a proband
Offspring of males with a mtDNA pathogenic variant will not inherit the variant.
All offspring of females with a mtDNA pathogenic variant are at risk of inheriting the pathogenic variant. If the female proband is heteroplasmic for the mtDNA pathogenic variant, the proportion of mutated mtDNA she transmits to offspring varies.
Clinical expression in offspring depends on the proportion of mutated mtDNA (mutational load), and the organs and tissues in which they are found (tissue distribution and threshold effect). Offspring often inherit different proportions of mutated mtDNA and therefore can have a wide range of clinical manifestations.
Other family members. The risk to other family members depends on the genetic status of the proband's mother: if she has a mtDNA pathogenic variant, her sibs and mother are also at risk.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
The parents of an affected child are obligate heterozygotes (i.e., presumed to be carriers of one pathogenic variant based on family history).
Molecular genetic testing for the pathogenic variants identified in the proband is recommended for the parents of the proband to confirm that both parents are heterozygous for a pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
If both parents are known to be heterozygous for a causative pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. Unless an affected individual's reproductive partner has (or is a carrier of) the same mitochondrial disorder, offspring will be obligate heterozygotes (carriers) for a pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a pathogenic variant.
Carrier detection. Carrier testing for at-risk relatives requires prior identification of the causative pathogenic variants in the family.
Autosomal Dominant Inheritance – Risk to Family Members
Parents of a proband
Some individuals diagnosed with an autosomal dominant mitochondrial disorder inherited a pathogenic variant from a heterozygous parent who may or may not have symptoms.
Alternatively, a proband may have the disorder as the result of a de novo pathogenic variant. The proportion of individuals with a mitochondrial disorder caused by a de novo pathogenic variant is unknown.
Molecular genetic testing for the pathogenic variant identified in the proband is recommended for the parents of the proband to confirm their genetic status and to allow reliable recurrence risk counseling.
If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
The proband has a de novo pathogenic variant.
The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.
The family history of an individual diagnosed with an autosomal dominant mitochondrial disorder may appear to be negative because of failure to recognize the disorder in family members, early death of the parent before the onset of symptoms, or late onset of the disease in the affected parent. Therefore, an apparently negative family history cannot be confirmed without appropriate clinical evaluation and/or molecular genetic testing (to establish that neither parent is heterozygous for the pathogenic variant identified in the proband).
Sibs of a proband. The risk to the sibs depends on the clinical/genetic status of the parents:
If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs is 50%.
If the proband has a known pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with an autosomal dominant mitochondrial disorder has a 50% chance of inheriting the causative pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the pathogenic variant, the parent's family members may be at risk.
X-Linked Inheritance – Risk to Family Members
Parents of a male proband
The father of an affected male will not have the disorder nor will he be hemizygous for the pathogenic variant; therefore, he does not require further evaluation/testing.
In a family with more than one affected individual, the mother of an affected male is an obligate heterozygote. Note: If a woman has more than one affected child and no other affected relatives and if the causative pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism.
If a male is the only affected family member (i.e., a simplex case), the mother may be a heterozygote (carrier), the affected male may have a de novo pathogenic variant (in which case the mother is not a carrier), or the mother may have somatic/germline mosaicism.
Molecular genetic testing of the mother is recommended to confirm her genetic status and to allow reliable recurrence risk assessment.
Sibs of a male proband. The risk to sibs depends on the genetic status of the mother:
If the mother of the proband has a pathogenic variant, the chance of transmitting it in each pregnancy is 50%. Males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant will be heterozygous may or may not be affected.
If the proband represents a simplex case and if the pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is presumed to be low but greater than that of the general population because of the possibility of maternal germline mosaicism.
Offspring of a male proband. Affected males transmit the pathogenic variant to all their daughters (who will be heterozygotes and may or may not be affected) and none of their sons.
Prenatal Testing
Mutation of mitochondrial DNA (mtDNA). Prenatal molecular genetic testing and interpretation for mtDNA disorders is difficult because of mtDNA heteroplasmy. The percentage level of mutated mtDNA in a chorionic villus sampling (CVS) biopsy may not reflect the percentage level of mutated mtDNA in other fetal tissues, and the percentage level may change during development and throughout life [Hellebrekers et al 2012].
The interpretation of a CVS result is difficult for most heteroplasmic mtDNA pathogenic variants. However, the variants m.8993T>G and m.8993T>C show a more even tissue distribution, and the percentage level of these two variants does not appear to change significantly over time. Successful prenatal molecular diagnosis has been carried out for pathogenic variants using DNA extracted from fetal cells obtained by amniocentesis (usually performed at ~15-18 weeks' gestation) or CVS (usually performed at ~10-12 weeks' gestation) [Hellebrekers et al 2012].
Mutation of nuclear DNA