Mitochondrial DNA Maintenance Defects Overview
Ayman W El-Hattab, MD, FAAP, FACMG, William J Craigen, MD, PhD, FACMG, Lee-Jun C Wong, PhD, FACMG, and Fernando Scaglia, MD, FAAP, FACMG.
Author Information and AffiliationsInitial Posting: March 8, 2018.
Estimated reading time: 15 minutes
Summary
This overview focuses on the clinical features and molecular genetics of mitochondrial DNA (mtDNA) maintenance defects.
The goals of this overview are the following.
Goal 2.
Review the genetic causes of mtDNA maintenance defects.
Goal 4.
Provide clinical and laboratory evaluation strategies to facilitate the diagnosis of a mtDNA maintenance defect and to establish a genetic cause in a proband (when possible).
Goal 5.
Summarize current management recommendations for individuals with mtDNA maintenance defects.
1. Mitochondrial DNA Maintenance Defects
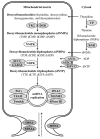
The maintenance of mtDNA is essential to the functioning of the mitochondria and, thus, to meeting the energy needs of all cells. The maintenance of mtDNA requires proteins essential for mtDNA synthesis, for maintenance of the mitochondrial nucleotide pool, and for mediating mitochondrial fusion [El-Hattab et al 2017].
Mitochondrial DNA is synthesized continuously and is not regulated by the cell cycle. The enzymes that synthesize mtDNA require a balanced supply of intramitochondrial nucleotides. These are supplied through mitochondrial nucleotide salvage pathways and the import of nucleotides from the cytosol via specific transporters. To function properly in mtDNA synthesis the quantities of these enzymes need to be perfectly balanced, a phenomenon achieved – in part – by the exchange of content between mitochondria through the process of mitochondrial fission and fusion.
The proteins known to be required for mtDNA synthesis are encoded by nuclear genes (i.e., genes found in the nucleus of cells). When pathogenic variants disrupt the function of any one of the proteins encoded by these genes, mtDNA synthesis is impaired, resulting in either quantitative defects in mtDNA (mtDNA depletion) or qualitative defects in mtDNA (multiple mtDNA deletions). These defects in mtDNA maintenance result in energy deficiency within cells. Cellular energy production insufficient to meet the needs of a given organ results in organ dysfunction (see ).
A diagram showing the proteins that are involved in mtDNA maintenance and known to be associated with mtDNA maintenance defects Enzymes of the mitochondrial nucleotide salvage pathway. Thymidine kinase 2 (TK2; encoded by TK2) and deoxyguanosine kinase (more...)
When first identified, defects in mtDNA maintenance were viewed as two clinically distinct groups of disorders:
Mitochondrial DNA depletion syndromes that typically present during infancy and are characterized by severe disease manifestations and shortened life expectancy; and
Multiple mtDNA deletion syndromes that typically present in adulthood and are characterized by milder disease manifestations including progressive external ophthalmoplegia (CPEO) and myopathy.
However, with the current understanding that both mtDNA depletion and multiple mtDNA deletions result from failure of proper mtDNA maintenance, it has become evident that these two groups of disorders represent the ends of a phenotypic continuum. The term "mtDNA maintenance defects" is used to represent the broad disease spectrum that encompasses both presentations as well as those that are intermediate.
2. Causes of mtDNA Maintenance Defects
To date, pathogenic variants in 20 nuclear genes are known to be associated with mtDNA maintenance defects. These genes and their primary presenting features are organized in Table 1 by the category of defect: mtDNA synthesis, mitochondrial nucleotide salvage pathway, cytosolic nucleotide metabolism, mitochondrial nucleotide import, and mitochondrial fusion.
Note: Disorders of mtDNA are not the subject of this overview (see Primary Mitochondrial Disorders Overview).
Table 1.
Categories of mtDNA Maintenance Defects: Genes and Primary Presenting Features
View in own window
Category
of Defect | Gene | Primary Presenting Features |
|---|
Encephalo-
hepatopathy | Encephalo-
myopathy | Encephalo-
neuropathy | Neurogastro-
intestinal
Encephalopathy | Myopathy | Ophthal-
moplegia | Optic
Atrophy | Neuropathy |
|---|
Mitochondrial
DNA
synthesis |
POLG
| X | X | X | X | X | X | | |
|
POLG2
| | | | | X | | | |
|
TWNK
| X | | X | | | X | | |
|
TFAM
| X | | | | | | | |
|
RNASEH1
| | X | | | | | | |
|
MGME1
| | | | | X | | | |
|
DNA2
| | | | | X | | | |
Mitochondrial
nucleotide
salvage
pathway |
TK2
| | | | | X | X | | |
|
DGUOK
| X | | | | X | | | |
|
SUCLA2
| | X | | | | | | |
|
SUCLG1
| | X | | | | | | |
|
ABAT
| | X | | | | | | |
Cytosolic
nucleotide
metabolism |
TYMP
| | | | X | | | | |
|
RRM2B
| | X | | X | | X | | |
Mitochondrial
nucleotide
import |
SLC25A4
| | | | | X | X | | |
|
AGK
| | | | | X | | | |
|
MPV17
| X | | | | | | | |
Mitochondrial
fusion |
OPA1
| | X | X | | | | X | |
|
MFN2
| | | | | | | X | X |
|
FBXL4
| | X | | | | | | |
3. Clinical Characteristics of mtDNA Maintenance Defects
Mitochondrial DNA maintenance defects are characterized by mtDNA depletion and/or multiple mtDNA deletions in mitochondria of cells of affected organs. The organs/tissues affected most often are the brain, liver, skeletal muscle, peripheral nerves, and gastrointestinal tract. Depending on the organ(s) predominantly affected, these disorders can be classified into groups associated mainly with encephalohepatopathy (see Table 2a), encephalomyopathy (Table 2b), encephaloneuropathy (Table 2c), neurogastrointestinal encephalopathy (Table 2d), myopathy (Table 2e), ophthalmoplegia (Table 2f), optic atrophy (Table 2g), or neuropathy (Table 2h).
Mitochondrial DNA Maintenance Defects Presenting with Encephalohepatopathy
Mitochondrial DNA maintenance defects manifesting as encephalohepatopathy (hepatocerebral) are typically associated with mtDNA depletion and generally present in neonates or infants with neurologic manifestations (including developmental delay and epilepsy), and with liver dysfunction and failure. Other common manifestations include growth failure, lactic acidosis, and hypoglycemia.
Table 2a.
Mitochondrial DNA Maintenance Defects Presenting with Encephalohepatopathy
View in own window
| Gene | Disorder/Phenotype | MOI | mtDNA
Maintenance
Defect | Usual Age
of Onset | Common Manifestations
in Addition to Liver
Dysfunction/Failure |
|---|
|
DGUOK
|
Deoxyguanosine kinase deficiency
| AR | Depletion | Neonatal | DD Hypotonia Nystagmus Lactic acidosis
|
|
MPV17
|
Hepatocerebral mtDNA depletion syndrome
| AR | Depletion | Neonatal
or infancy | DD Hypotonia Failure to thrive Hearing impairment Lactic acidosis
|
|
POLG
|
Alpers-Huttenlocher syndrome
| AR | Depletion | Early
childhood | DD Psychomotor regression Epilepsy Hearing impairment
|
|
TFAM
| Encephalohepatopathy (OMIM 617156) | AR | Depletion | Neonatal |
|
|
TWNK
| Encephalohepatopathy (OMIM 271245) | AR | Depletion | Neonatal
or infancy | DD Hypotonia Lactic acidosis
|
AR = autosomal recessive; DD = developmental delay; IUGR = intrauterine growth restriction; MOI = mode of inheritance; mtDNA = mitochondrial DNA
Mitochondrial DNA Maintenance Defects Presenting with Encephalomyopathy
The majority of encephalomyopathic mtDNA maintenance defects are associated with mtDNA depletion and are early-onset diseases with an infantile presentation. The two disorders, however, that are usually associated with multiple mtDNA deletions rather than depletion are adult-onset diseases: POLG-related myoclonic epilepsy-myopathy-sensory ataxia and RNASEH1-related encephalomyopathy.
Table 2b.
Mitochondrial DNA Maintenance Defects Presenting with Encephalomyopathy
View in own window
AR = autosomal recessive; CSF = cerebrospinal fluid; DD = developmental delay; GI = gastrointestinal; MOI = mode of inheritance; mtDNA = mitochondrial DNA
Mitochondrial DNA Maintenance Defects Presenting with Encephaloneuropathy
Mitochondrial DNA maintenance defects exhibiting encephaloneuropathy can be associated with mtDNA depletion or multiple mtDNA deletions, and are characterized by manifestations related to the central and peripheral nervous systems.
Table 2c.
Mitochondrial DNA Maintenance Defects Presenting with Encephaloneuropathy
View in own window
| Gene | Disorder | MOI | mtDNA Maintenance
Defect | Usual Age
of Onset | Common Manifestations in
Addition to Peripheral
Neuropathy & Ataxia |
|---|
|
OPA1
| Behr syndrome
(OMIM 210000) | AR | NA | Infancy or
early childhood | Vision impairment Optic nerve pallor
|
|
POLG
|
Ataxia neuropathy spectrum disorders
| AR | Multiple deletions | Early adulthood | Epilepsy |
|
TWNK
| Infantile-onset spinocerebellar ataxia (OMIM 271245) | AR | Depletion | 2nd year of life | Hypotonia Hearing impairment
|
AR = autosomal recessive; MOI = mode of inheritance; mtDNA = mitochondrial DNA
Mitochondrial DNA Maintenance Defects Presenting with Neurogastrointestinal Encephalopathy
Mitochondrial neurogastrointestinal encephalopathy (MNGIE) is characterized by a variable age of onset (generally in the 2nd decade) and progressive gastrointestinal dysmotility, peripheral neuropathy, and leukoencephalopathy. The gastrointestinal manifestations of the disease may mimic anorexia nervosa. MNGIE is most commonly caused by biallelic pathogenic variants in TYMP, the gene encoding thymidine phosphorylase; however, biallelic pathogenic variants in POLG or RRM2B also cause this disorder.
Table 2d.
Mitochondrial DNA Maintenance Defects Presenting with Neurogastrointestinal Encephalopathy
View in own window
| Gene | Disorder | MOI | mtDNA Maintenance
Defect | Usual Age of Onset | Common Manifestations |
|---|
|
TYMP
|
MNGIE type 1
| AR | Depletion & multiple deletions | Adolescence
or early adulthood | GI dysmotility Cachexia Peripheral neuropathy Ophthalmoplegia Muscle weakness Leukoencephalopathy 1
|
|
POLG
|
MNGIE type 4B
| AR | Depletion & multiple deletions | Infancy or childhood |
|
RRM2B
|
MNGIE type 8B
| AR | Depletion | Early adulthood |
AR = autosomal recessive ; GI = gastrointestinal; MOI = mode of inheritance; mtDNA = mitochondrial DNA
- 1.
Note: Leukoencephalopathy is not present in POLG-related neurogastrointestinal encephalopathy.
Mitochondrial DNA Maintenance Defects Presenting with Myopathy
Myopathic mtDNA maintenance defects include a group of diseases that vary in their age of onset. Skeletal muscles are the main system involved in all of them. Cardiomyopathy can occur in some of these disorders.
Table 2e.
Mitochondrial DNA Maintenance Defects Presenting with Myopathy
View in own window
| Gene | Disorder | MOI | mtDNA
Maintenance
Defect | Usual Age
of Onset | Common Clinical Manifestations
in Addition to Muscle Weakness |
|---|
|
AGK
| Sengers syndrome
(OMIM 212350) | AR | Depletion | Neonatal period |
|
|
DGUOK
|
Myopathy
| AR | Multiple deletions | Early or mid-
adulthood |
|
|
DNA2
| Myopathy
(OMIM 615156) | AD | Multiple deletions | Childhood or
early adulthood |
|
|
MGME1
| Myopathy
(OMIM 615084) | AR | Depletion & multiple deletions | Childhood or
early adulthood |
|
|
POLG2
| Myopathy
(OMIM 610131) | AD | Multiple deletions | Infancy to
adulthood |
|
|
SLC25A4
| Cardiomyopathy
(OMIM 615418) | AR | Multiple deletions | Childhood |
|
Cardiomyopathy
(OMIM 617184) | AD | Depletion | Birth |
|
|
TK2
|
Mitochondrial DNA depletion syndrome
| AR | Depletion | Infancy or
childhood |
|
AD = autosomal dominant; AR = autosomal recessive; MOI = mode of inheritance; mtDNA = mitochondrial DNA
Mitochondrial DNA Maintenance Defects Presenting with Ophthalmoplegia
Mitochondrial DNA maintenance defects that cause ophthalmoplegia are associated with multiple DNA deletions and are characterized by progressive weakness of the extraocular eye muscles resulting in ptosis (drooping of the eyelids) and ophthalmoplegia (paralysis of the extraocular muscles causing limitation in horizontal and vertical eye movements).
Although these are typically diseases of adulthood, earlier onset can be seen in the recessively inherited diseases. Although ophthalmoplegia and ptosis are consistent, and are the main manifestations in these diseases, a more generalized myopathy (sometimes mild) can be observed in some affected individuals.
Table 2f.
Mitochondrial DNA Maintenance Defects Presenting with Ophthalmoplegia
View in own window
| Gene | Disorder | MOI | mtDNA
Maintenance
Defect | Usual Age
of Onset | Common Clinical Manifestations
in Addition to Ptosis & Ophthalmoplegia |
|---|
|
POLG
|
Progressive external ophthalmoplegia
| AR | Multiple deletions | Adolescence
or young
adulthood | Easy fatigability / exercise intolerance |
| AD | Multiple deletions | Adulthood | Easy fatigability / exercise intolerance |
|
RRM2B
|
Chronic progressive external ophthalmoplegia
| AR | Multiple deletions | Childhood | Muscle weakness Bulbar dysfunction
|
| AD | Multiple deletions | Adulthood | Ataxia Muscle weakness Bulbar dysfunction
|
|
SLC25A4
| Progressive external ophthalmoplegia
(OMIM 609283) | AD | Multiple deletions | Adulthood | Easy fatigability / exercise intolerance |
|
TK2
| Progressive external ophthalmoplegia
(OMIM 617069) | AR | Multiple deletions | Adulthood | Muscle weakness |
|
TWNK
| Progressive external ophthalmoplegia
(OMIM 609286) | AD | Multiple deletions | Early
adulthood | Easy fatigability / exercise intolerance |
AD = autosomal dominant; AR = autosomal recessive; MOI = mode of inheritance; mtDNA = mitochondrial DNA
Mitochondrial DNA Maintenance Defects Presenting with Optic Atrophy
Table 2g.
Mitochondrial DNA Maintenance Defects Presenting with Optic Atrophy
View in own window
| Gene | Disorder | MOI | mtDNA Maintenance
Defect | Usual Age
of Onset | Common Clinical Manifestations |
|---|
|
OPA1
| Optic atrophy type 1 (OMIM 165500) | AD | Multiple deletions | Childhood | Vision impairment |
|
MFN2
| Optic atrophy | AD | Multiple deletions | Early childhood | Vision impairment Optic nerve pallor Peripheral neuropathy Muscle weakness
|
AD = autosomal dominant; MOI = mode of inheritance
Mitochondrial DNA Maintenance Defects Presenting with Neuropathy
Table 2h.
Mitochondrial DNA Maintenance Defects Presenting with Neuropathy
View in own window
AD = autosomal dominant; MOI = mode of inheritance; mtDNA = mitochondrial DNA; NA= not available
4. Evaluation Strategies to Diagnose mtDNA Maintenance Defects and to Establish a Genetic Cause in a Proband
Establishing a specific genetic cause of a mtDNA maintenance defect can aid in discussion of prognosis (which is beyond the scope of this GeneReview) and in genetic counseling (see Genetic Counseling).
Establishing the specific genetic cause of a mtDNA maintenance defect usually requires a medical history, physical and neurologic examination, laboratory testing including routine studies and specialized biochemical genetic studies, imaging studies such as brain MRI, echocardiogram, abdominal ultrasound examination, family history, and genomic/genetic testing.
Clinical and Laboratory Findings
The diagnosis of a mtDNA maintenance defect is suspected based on the involved organs, age of onset, and results of commonly available laboratory tests (e.g., presence of lactic acidemia or methylmalonic aciduria).
Biopsies of affected tissues typically show mtDNA depletion and/or multiple mtDNA deletions as well as decreased activity of multiple electron transport complexes (ETC). However, because tissue biopsies are often invasive procedures, molecular genetic testing of leukocyte DNA is typically performed first to determine if the diagnosis of a mtDNA maintenance defect can be established. When molecular genetic test results are equivocal or fail to confirm the diagnosis of a mtDNA maintenance defect, tissue samples can be obtained to assay for mtDNA depletion and/or multiple mtDNA deletions and to assess ETC activity.
Family History
A three-generation family history should be obtained, with attention to relatives with signs and symptoms that could be related to a mtDNA maintenance defect and documentation of relevant findings through direct physical examination, or review of medical records, including results of molecular genetic testing.
Molecular Genetic Testing
Approaches include gene-targeted testing (multigene panel, single-gene testing) or comprehensive genomic testing (exome sequencing). Gene-targeted testing requires the clinician to hypothesize which gene(s) are likely involved, whereas genomic testing may not. Options for testing include the following:
5. Management of Individuals with mtDNA Maintenance Defects
Most mtDNA maintenance defects affect multiple organs; therefore, affected individuals need comprehensive evaluations to assess the degree of involvement of different organs. Management should also involve a multidisciplinary team to provide clinical care for these multiorgan diseases.
Assessment of Disease Extent
For individuals with chronic disease, Table 3 summarizes evaluations that are recommended if they have not already been completed:
Table 3.
Recommended Evaluations Following Initial Diagnosis in a Proband with a mtDNA Maintenance Defect Resulting in Chronic Disease
View in own window
System/
Concern | Evaluation | Comment |
|---|
|
Eyes
| Ophthalmologic eval (See POLG-Related Disorders.) | To assess for optic atrophy, ptosis, ophthalmoplegia, & nystagmus |
|
ENT/Mouth
| Hearing eval (See POLG-Related Disorders.) | |
|
Cardiovascular
| Echocardiogram Electrocardiogram
| For persons w/myopathy |
|
Respiratory
|
| To identify respiratory insufficiency in persons w/myopathy |
|
Gastrointestinal
|
| For persons w/hepatopathy |
For MNGIE disease (see Mitochondrial Neurogastrointestinal Encephalopathy Disease):
Consultation w/gastroenterologist Depending on manifestations: abdominal films, abdominal CT, upper GI contrast radiography, esophagogastroduodenoscopy, sigmoidoscopy, liquid phase scintigraphy, antroduodenal manometry
| To evaluate for gastrointestinal dysmotility |
|
Feeding
| Swallowing assessment Nutritional eval
| In individuals w/feeding difficulty & growth failure 1 |
|
Renal
|
| To evaluate for tubulopathy |
|
Neurologic
| Comprehensive neurologic exam Brain MRI & MRS Nerve conduction studies & electromyography (if neuropathy is suspected) Electroencephalography (if seizures are suspected)
| For persons w/neurologic manifestations |
|
Musculoskeletal
| Referral to rehabilitation specialist | Evaluate gait, weakness, safety, activities of daily living |
|
Genetics & metabolic
| Consultation w/clinical geneticist &/or genetic counselor Lactate level to evaluate for lactic acidosis Glucose level to evaluate for hypoglycemia
| |
Treatment of Manifestations
Currently there is no clinical therapy to treat the primary defect in affected individuals. Management, which is primarily supportive, is outlined in Table 4. Some specific considerations:
As exogenous thymidine phosphorylase can improve outcome in MNGIE resulting from thymidine phosphorylase deficiency, experimental therapy for MNGIE includes both bone marrow and liver transplantation.
Nucleoside therapy has been considered in TK2 deficiency.
Affected individuals may be at increased risk for acidosis and hypoglycemia during illness and surgery and protocols to prevent prolonged fasting should be provided.
Table 4.
Treatment of Manifestations in a Proband with a mtDNA Maintenance Defect Resulting in Chronic Disease
View in own window
Manifestation/
Concern | Treatment |
|---|
|
Ptosis
| Ptosis blepharoplasty |
|
Sensorineural hearing loss
| Hearing aids & cochlear implantation (See POLG-Related Disorders, RRM2B Mitochondrial DNA Maintenance Defects, Genetic Hearing Loss Overview.) |
|
Cardiomyopathy, hypertrophic or dilated
| Referral to cardiologist Standard treatment
|
|
Respiratory insufficiency
|
|
|
Liver failure
| Referral to hepatologist Reduction in dietary protein Correction of coagulopathy Frequent or continuous feeding to prevent hypoglycemia Consideration of liver transplant
|
|
Gastrointestinal dysmotility
| Referral to gastroenterologist Nutritional support Total parenteral nutrition Domperidone Antibiotic therapy for intestinal bacterial overgrowth
|
Failure to thrive &
feeding difficulties
|
|
|
Renal tubulopathy
|
|
|
Neuropathy
|
|
|
Seizures
|
|
|
Hypoglycemia
|
|
ASM = anti-seizure medication
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy. In the US, early intervention is a federally funded program available in all states.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed.
Ages 5-21 years
In the US, an IEP based on the individual's level of function should be developed by the local public school district. Affected children are permitted to remain in the public school district until age 21.
Discussion about transition plans including financial, vocation/employment, guardianship, and medical arrangements should begin at age 12 years. Developmental pediatricians can provide assistance with transition to adulthood.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies and to support parents in maximizing quality of life.
Consideration of private supportive therapies based on the affected individual's needs is recommended. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
In the US:
Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
Physical therapy is recommended to maximize mobility.
Consider use of durable medical equipment as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction. Assuming that it is safe for the individual to eat by mouth, feeding therapy, typically from an occupational or speech therapist, is recommended for affected individuals who have difficulty feeding as a result of poor oral motor control.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties.
6. Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Mitochondrial DNA maintenance defects can be inherited in an autosomal recessive or autosomal dominant manner.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
The parents of an affected child are obligate heterozygotes (i.e., carriers of one mtDNA maintenance defect-related pathogenic variant).
Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. Offspring of an individual with a mtDNA maintenance defect are obligate heterozygotes (carriers) for a pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a mtDNA maintenance defect-related pathogenic variant.
Carrier detection. Carrier testing for at-risk relatives requires prior identification of the pathogenic variants in the family.
Autosomal Dominant Inheritance – Risk to Family Members
Parents of a proband
Most individuals diagnosed with a mtDNA maintenance defect have an affected parent.
Some individuals diagnosed with a mtDNA maintenance defect have the disorder as the result of a de novo pathogenic variant.
Molecular genetic testing is recommended for the parents of a proband with an apparent de novo pathogenic variant.
If the pathogenic variant found in the proband cannot be detected in leukocyte DNA of either parent, possible explanations include a de novo pathogenic variant in the proband or germline mosaicism in a parent. Though theoretically possible, no instances of germline mosaicism have been reported to date.
The family history of some individuals diagnosed with a mtDNA maintenance defect may appear to be negative because of failure to recognize the disorder in family members, early death of the parent before the onset of symptoms, or late onset of the disease in the affected parent. Therefore, an apparently negative family history is not definitive unless appropriate clinical evaluation and/or molecular genetic testing has been performed for the parents of the proband.
Sibs of a proband
The risk to sibs of the proband depends on the genetic status of the proband's parents.
If a parent of the proband is affected and/or is heterozygous for the pathogenic variant identified in the proband, the risk to sibs is 50%.
If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [
Rahbari et al 2016].
If the parents have not been tested for the mtDNA maintenance defect-related pathogenic variant but are clinically unaffected, the risk to sibs of a proband appears to be low.
Offspring of a proband. Each child of an individual with a mtDNA maintenance defect has a 50% chance of inheriting the pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the mtDNA maintenance defect-related pathogenic variant, the parent's family members may be at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the mtDNA maintenance defect-related pathogenic variant(s) have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal and preimplantation genetic testing. While most health care professionals would consider use of prenatal and preimplantation genetic testing to be a personal decision, discussion of these issues may be helpful.
References
Literature Cited
El-Hattab, AW, Craigen
WJ, Scaglia
F. Mitochondrial DNA maintenance defects.
Biochim Biophys Acta Mol Basis Dis.
2017;1863:1539-55.
[
PubMed: 28215579]
Rahbari
R, Wuster
A, Lindsay
SJ, Hardwick
RJ, Alexandrov
LB, Turki
SA, Dominiczak
A, Morris
A, Porteous
D, Smith
B, Stratton
MR, Hurles
ME, et al.
Timing, rates and spectra of human germline mutation.
Nat Genet.
2016;48:126-33.
[
PMC free article: PMC4731925] [
PubMed: 26656846]