Summary
Clinical characteristics.
COL1A1/2 osteogenesis imperfecta (COL1A1/2-OI) is characterized by fractures with minimal or absent trauma, variable dentinogenesis imperfecta (DI), and, in adult years, hearing loss. The clinical features of COL1A1/2-OI represent a continuum ranging from perinatal lethality to individuals with severe skeletal deformities, mobility impairments, and very short stature to nearly asymptomatic individuals with a mild predisposition to fractures, normal dentition, normal stature, and normal life span. Fractures can occur in any bone but are most common in the extremities. DI is characterized by gray or brown teeth that may appear translucent, wear down, and break easily. COL1A1/2-OI has been classified into four types based on clinical presentation and radiographic findings. This classification system can be helpful in providing information about prognosis and management for a given individual. The four more common OI types are now referred to as follows:
- Classic non-deforming OI with blue sclerae (previously OI type I)
- Perinatally lethal OI (previously OI type II)
- Progressively deforming OI (previously OI type III)
- Common variable OI with normal sclerae (previously OI type IV)
Diagnosis/testing.
The diagnosis of COL1A1/2-OI is established in a proband by identification of a heterozygous pathogenic or likely pathogenic variant in COL1A1 or COL1A2 by molecular genetic testing.
Management.
Treatment of manifestations: Ideally, management is by a multidisciplinary team including specialists in medical management of OI, clinical genetics, orthopedics, rehabilitation medicine, pediatric dentistry, otology/otolaryngology, and mental health. Parents / other caregivers must practice safe handling techniques. Mainstays of treatment include: bracing of limbs depending on OI severity; orthotics to stabilize lax joints; physical activity; physical and occupational therapy to maximize bone stability, improve mobility, prevent contractures, prevent head and spine deformity, and improve muscle strengthening; mobility devices as needed; and pain management. Fractures are treated with: as short a period of immobility as is practical; small and lightweight casts; physical therapy as soon as casts are removed; and intramedullary rodding when indicated to provide anatomic positioning of limbs. Progressive scoliosis in severe OI may not respond well to conservative or surgical management. Bisphosphonates continue to be used most extensively in severely affected children with OI. Surgical treatment for basilar impression should be done in a center experienced in the necessary procedures. Dental care strives to maintain both primary and permanent dentition, a functional bite or occlusion, optimal gingival health, and overall appearance. Conductive hearing loss may be improved with middle ear surgery; later-onset sensorineural hearing loss is treated in the same manner as when caused by other conditions. Mental health support through psychiatry/psychology and appropriate social worker intervention can improve quality of life.
Prevention of secondary complications: During general anesthesia, proper positioning on the operating room table and use of cushioning such as egg crate foam can help avoid fractures.
Surveillance: Orthopedic evaluation with ancillary therapy services (physical and rehabilitation medicine) as indicated every three months until age one year, every six months from ages one to three years, and then annually or with any new fractures. Physical therapy evaluation in infancy for those with motor delays and as needed to improve mobility and function. CT and/or MRI examination with views across the base of the skull to evaluate for basilar impression if concerning signs or symptoms are present. Cervical spine flexion and extension radiographs in children able to cooperate with the examination or before participating in sporting activities in more mildly affected individuals. Twice-yearly dental visits beginning in early childhood or even infancy for those with (or at risk for) DI. Hearing evaluation at three- to five-year intervals from age five years until hearing loss is identified, then as indicated based on the nature and degree of hearing loss and associated interventions.
Agents/circumstances to be avoided: Contact sports should be avoided.
Genetic counseling.
COL1A1/2-OI is inherited in an autosomal dominant manner. The proportion of affected individuals who represent simplex cases (i.e., a single occurrence of the disorder in a family) varies by the severity of disease. Approximately 60% of probands with mild OI represent simplex cases. Virtually 100% of probands with progressively deforming or perinatally lethal OI represent simplex cases and have a de novo pathogenic variant or a pathogenic variant inherited from a parent with somatic and/or germline mosaicism. Parental somatic and/or germline mosaicism is present in up to 16% of families. Each child of an individual with a dominantly inherited form of COL1A1/2-OI has a 50% chance of inheriting the causative variant and of developing some manifestations of OI. Prenatal testing in at-risk pregnancies can be performed by molecular genetic testing if the COL1A1 or COL1A2 causative variant has been identified in an affected relative. Ultrasound examination performed in a center with experience in diagnosing OI can be valuable in the prenatal diagnosis of the lethal form and most severe forms prior to 20 weeks' gestation; milder forms may be detected later in pregnancy if fractures or deformities occur.
Diagnosis
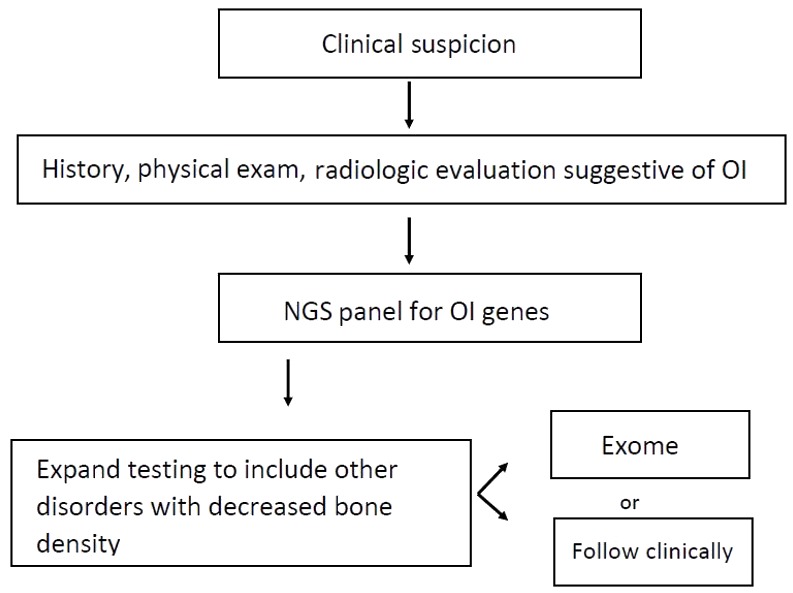
An algorithm for the diagnosis of osteogenesis imperfecta (OI) has been published [Basel & Steiner 2009]. See Figure 1.
Figure 1.
Recommended testing algorithm for evaluation of osteogenesis imperfecta Adapted from Basel & Steiner [2009]
Suggestive Findings
COL1A1/2 osteogenesis imperfecta (OI) should be suspected in individuals with the following clinical, radiographic, and laboratory features.
Clinical features (Table 1)
- Fractures with minimal or no trauma in the absence of other factors, such as non-accidental trauma (NAT) or other known disorders of bone
- Short stature or stature shorter than predicted based on stature of unaffected family members, often with bone deformity
- Blue/gray scleral hue
- Dentinogenesis imperfecta (DI)
- Progressive, postpubertal hearing loss
- Ligamentous laxity and other signs of connective tissue abnormality
- Family history of OI, usually consistent with autosomal dominant inheritance
Table 1.
Clinical Features of COL1A1/2 Osteogenesis Imperfecta by Type
Radiographic features of OI change with age. The major findings include the following (Table 2):
- Fractures of varying ages and stages of healing, often of the long bones but may also rarely involve ribs and skull. Metaphyseal fractures can be seen in a very small number of children with OI. Rib fractures are much more common in NAT than in OI.
- "Codfish" vertebrae, which are the consequence of spinal compression fractures, seen more commonly in adults
- Wormian bones, defined as "sutural bones which are 6 mm by 4 mm (in diameter) or larger, in excess of ten in number, with a tendency to arrangement in a mosaic pattern" [Cremin et al 1982]. Wormian bones are suggestive of but not pathognomonic for OI.
- Protrusio acetabuli, in which the socket of the hip joint is too deep and the acetabulum bulges into the cavity of the pelvis causing intrapelvic protrusion of the acetabulum
- Low bone mass or osteoporosis detected by dual-energy x-ray absorptiometry (DXA). Bone density can be normal, especially in individuals with OI type I, as DXA measures mineral content rather than collagen [Deodhar & Woolf 1994, Paterson & Mole 1994, Cepollaro et al 1999, Lund et al 1999].Note: (1) A major determinant of bone density may be the individual's ability to ambulate. (2) Bone density standards for children under age two years have been determined after sampling very small populations (often <10 persons); thus, reliability is an issue. (3) Bone density standards for children are based on height; corrections for short stature of severely affected individuals need to be made. (4) Bone density is not typically measured in children before age four years because of their inability to lie still, though this may be accomplished with patience in sleeping infants. (5) The purpose of measuring bone density in individuals known to have OI is to allow for monitoring of the individual's bone density over time, and not for comparison with unaffected individuals.
Table 2.
Radiographic Findings of COL1A1/2 Osteogenesis Imperfecta by Type
Laboratory features
- Serum concentrations of vitamin D, calcium, phosphorous, and alkaline phosphatase are typically normal; however, alkaline phosphatase may be elevated acutely in response to fracture and rare instances of abnormally low alkaline phosphatase levels have been noted anecdotally in severe OI.
- Analysis of type 1 collagen synthesized in vitro by culturing dermal fibroblasts obtained from a small skin biopsy reflects the structure and quantity of the collagen. The sensitivity of biochemical testing is approximately 90% in individuals with clinically confirmed OI [Wenstrup et al 1990; PH Byers, personal communication]. Biochemical analysis is essentially no longer used clinically with the advances in molecular diagnostics.
Establishing the Diagnosis
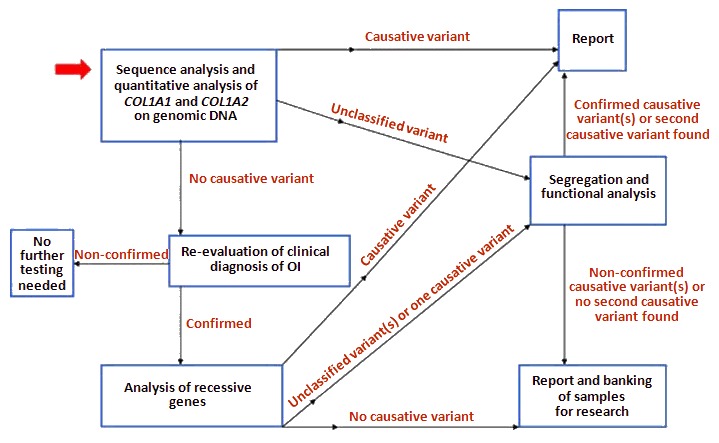
The diagnosis of COL1A1/2-OI is established in a proband by identification of a heterozygous pathogenic (or likely pathogenic) variant in COL1A1 or COL1A2 by molecular genetic testing (see Table 3). An approach to the molecular diagnosis of OI has been published (see Figure 2) [van Dijk et al 2012], but such approaches are in flux as technology is changing rapidly.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of a heterozygous COL1A1 or COL1A2 variant of uncertain significance does not establish or rule out the diagnosis
Molecular genetic testing approaches can include a combination of gene-targeted testing (concurrent gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of COL1A1/2-OI, molecular genetic testing approaches can include concurrent gene testing or use of a multigene panel:
- Concurrent gene testing. Sequence analysis of COL1A1 and COL1A2 detects missense, nonsense, and splice site variants and small intragenic deletions/insertions; typically, exon or whole-gene deletions/duplications are not detected. Perform sequence analysis first. If no pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications.
- A multigene panel that includes COL1A1, COL1A2, and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder, a multigene panel that also includes deletion/duplication analysis is recommended (see Table 3).
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by bone fragility and/or skeletal dysplasia, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible and becoming more widely available.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 3.
Molecular Genetic Testing Used in COL1A1/2 Osteogenesis Imperfecta
Clinical Characteristics
Clinical Description
The severity of COL1A1/2 osteogenesis imperfecta (COL1A1/2-OI) ranges from perinatal lethality to individuals with severe skeletal deformities, mobility impairments, and very short stature to nearly asymptomatic individuals with a mild predisposition to fractures, normal stature, and normal life span.
COL1A1/2-OI is classified into four more common types based on clinical presentation, radiographic features, family history, and natural history [Sillence et al 1979]. An update of the Sillence classification has been proposed and has gained some acceptance [Emery & Rimoin 2012]. Although this classification of COL1A1/2-OI into types is helpful in providing information about prognosis and management of a given individual, the features of different types of COL1A1/2-OI overlap and it is not always easy to categorize the extent of the clinical disorder. It is helpful to remember that the severity of clinical and radiographic features lies on a continuum and that the "types" are defined using characteristics that appear to form clinical "nodes." Interfamilial variability is apparent among individuals with the same OI type and intrafamilial variability is apparent among individuals with the same causative variant. Nonetheless, it is reasonable to continue to think of COL1A1/2-OI in terms of these types in order to provide information about the expected natural history of the disorder.
Classic non-deforming OI with blue sclerae (previously OI type I) is characterized by blue sclerae and normal stature. A small proportion of infants with OI type I have femoral bowing at birth. The first fractures may occur at birth or with diapering. More often, the first fractures occur when the infant begins to walk and, more importantly, to fall. Fractures generally occur at a rate of a few to several per year and then decrease in frequency after puberty. Fracture frequency often increases again in adulthood, especially in postmenopausal women and men beyond the fifth decade [Paterson et al 1984]. Affected individuals may have anywhere from a few fractures to more than 100, but the fractures usually heal normally with no resulting deformity.
Most affected individuals have normal or near normal stature but are often shorter than other members of their families and shorter than predicted based on parental heights.
Joint hypermobility predisposes to a number of minor comorbidities. The primary clinical concern is early-onset degenerative joint disease due to malalignment of articular surfaces.
In their classification of OI, Sillence et al [1979] designated a subset of classic non-deforming OI with dentinogenesis imperfecta (DI) (OI type IB). In individuals with DI, morbidity results not from dental decay but rather from premature wearing down of the teeth. DI can be a significant cosmetic concern. Dental eruption in classic non-deforming OI can sometimes occur early.
Progressive hearing loss occurs in about 50% of adults with classic non-deforming OI, beginning as a conductive hearing loss but often with an additional sensorineural hearing loss component in time.
Perinatally lethal OI (previously OI type II). Abnormalities characteristic of perinatally lethal OI are evident at birth. Weight and length are small for gestational age. The sclerae are dark blue and connective tissue is extremely fragile. The skull is large for the body size and soft to palpation. Callus formation on the ribs may be palpable. Extremities are short and bowed. Hips are usually flexed and abducted in a "frog-leg" position. Although some fetuses with perinatally lethal OI die in utero or are spontaneously aborted, more typically infants die in the immediate perinatal period. More than 60% of affected infants die on the first day; 80% die within the first week; survival beyond one year is exceedingly rare and usually involves intensive support such as continuous assisted ventilation [Byers et al 1988]. Death usually results from pulmonary insufficiency related to the small thorax, rib fractures, or flail chest because of unstable ribs. Those who survive the first few days of life may not be able to ingest sufficient calories because of respiratory distress.
Histologic evaluation of bone from infants with perinatally lethal OI shows marked reduction in collagen in secondary trabeculae and cortical bone [Horton et al 1980]. Cortical bone is hypercellular with large osteocytes. Trabeculae contain woven bone with large immature osteoblasts [Cole et al 1992, Cole & Dalgleish 1995].
Progressively deforming OI (previously OI type III). The diagnosis of progressively deforming OI is readily apparent at birth. Fractures in the newborn period, simply with handling of the infant, are common. In some affected infants, the number and severity of rib fractures lead to death from pulmonary failure in the first few weeks or months of life.
Infants who survive this period generally fare well, although most do not walk without assistance and usually use a wheelchair or other assistance for mobility because of severe bone fragility and marked bone deformity. Affected individuals have as many as 200 fractures and progressive deformity even in the absence of obvious fracture. Progressively deforming OI is often difficult to manage orthopedically, even with intramedullary rod placement.
Growth is extremely delayed and adults with progressively deforming OI are among the shortest individuals known, with some having adult stature of less than one meter.
Intellect is normal unless there have been intracerebral hemorrhages (extremely rare). Faqeih et al [2009] published a report identifying increased risk for intracranial hemorrhage (ICH) in a "small number" of individuals who were identified to have pathogenic variants affecting exon 49 of COL1A2, which codes for the most carboxy-terminal part of the triple-helical domain of the collagen alpha-2(I) chain. They concluded that this pathogenic variant appeared to increase the risk for abnormal limb development and intracranial bleeding. Budsamongkol et al [2019] reported a young boy with marked joint hypermobility, significant DI, brachydactyly, and a COL1A2 pathogenic variant found to be associated with ICH by Faqeih et al [2009]. The boy had not experienced an ICH, but as some of the original affected individuals only presented with ICH in their teenage years, this does not eliminate the risk in this young individual.
Even within progressively deforming OI, considerable heterogeneity is observed at the clinical level. Some individuals have normal-appearing teeth and facies while others have DI, a large head, and enlarged ventricles that reflect the soft calvarium. Relative macrocephaly and barrel chest deformity are observed. Usually sclerae are blue in infancy but lighten with age. Hearing loss generally begins in the teenage years. As molecular testing of this subgroup further differentiates those with COL1A1/2-OI from the autosomal recessive forms, the clinical profile of this heterogeneous group will become more refined.
Basilar impression, an abnormality of the craniovertebral junction caused by descent of the skull on the cervical spine, is common. Basilar impression is characterized by invagination of the margins of the foramen magnum upward into the skull, resulting in protrusion of the odontoid process into the foramen magnum. Basilar impression may progress to brain stem compression, obstructive hydrocephalus, or syringomyelia because of direct mechanical blockage of normal CSF flow [Charnas & Marini 1993, Sillence 1994, Hayes et al 1999]. Symptoms of basilar impression become apparent with neck flexion. Findings include posterior skull pain, C2 sensory deficit, tingling in the fourth and fifth digits, and numbness in the medial forearm. When swimming, affected individuals may perceive that water temperature differs below and above the umbilicus. Lhermitte's sign (tingling on neck flexion) can be demonstrated at any stage. Basilar impression can cause headache with coughing, trigeminal neuralgia, loss of function of the extremities, or paresthesias. At its most severe involvement, sleep apnea and death can occur.
Common variable OI with normal sclerae (previously OI type IV) is characterized by mild short stature, DI, adult-onset hearing loss, and normal-to-gray sclerae. This is the most variable form of OI, ranging in severity from moderately severe to so mild that it may be difficult to make the diagnosis.
Stature is variable and may vary markedly within the family. DI is common but may be mild. Sclerae are typically light blue or gray at birth but quickly lighten to near normal. Hearing loss occurs in some and basilar impression can occur.
Other Considerations
Facial features. Infants and children with OI are often described as having a triangular face. The skull is relatively large compared to body size.
Other skeletal problems. Individuals with OI may also have scoliosis, early-onset arthritis, non-inflammatory arthralgia, and myofascial pain.
Skin. Easy bruising is a frequent observation in individuals with OI. This is believed to be caused by microvascular fragility and poor microstructural support of the connective tissues.
Hearing loss. Mixed conductive and sensorineural hearing loss afflicts the majority of adults with OI. Childhood-onset hearing loss affects approximately 7% of affected children between ages five and nine years; progressive postpubertal hearing loss is more typical. The initial conductive hearing loss results from fractures of the bones of the middle ear with contracture and scarring of the incus. With age, sensorineural hearing loss compounds the preexisting conductive element. Fixation of the stapes is not unlike otosclerosis and surgical techniques such as stapedotomy used to treat otosclerosis have shown similar success in treating hearing loss in OI [van der Rijt & Cremers 2003, Kuurila et al 2004, Doi et al 2007]. Bisphosphonate therapy has not been shown to influence hearing loss.
Gastrointestinal. Although complaints of constipation are common in adults with OI who are mobile in wheelchairs, it is not clear if this is a complication of OI itself or of the mode of transport. Bowel obstruction can occur as a result of protrusio acetabuli [Lee et al 1995] but appears to be uncommon.
Cardiovascular. Emerging data support an increased risk for cardiac and vascular disease in OI. Ashournia et al [2015] performed a systematic review of the literature in 2015 documenting a broad array of cardiovascular phenotypes with higher prevalence in individuals with a clinical diagnosis of OI including arterial and aortic dissection. Balasubramanian et al [2019] reported three additional individuals with COL1A1/2-OI and aortic aneurysms. There is still no consensus on cardiovascular surveillance, although some centers have initiated screening echocardiograms every three to five years to monitor for this risk.
Development. Cognition is expected to be normal but gross motor development may be hindered by joint hypermobility and progressive deformity due to recurrent fractures.
Functional limitations. Individuals with OI may experience other functional limitations, although these will be highly dependent on the specific physical manifestations of OI.
Life expectancy. The severely affected neonates with perinatally lethal OI typically do not survive, with a significant proportion of infants dying within the first 48 hours. Aggressive life support can prolong survival but ultimately the most severe forms remain perinatally lethal. Life expectancy for classic non-deforming OI and common variable OI is normal. Progressively deforming OI is highly variable and life expectancy may be shortened by the presence of severe kyphoscoliosis with attendant restrictive pulmonary disease resulting in cardiac insufficiency.
Phenotype Correlations by Gene
Most commonly OI results from pathogenic heterozygous variants in either of the genes encoding the alpha helical chains of type 1 collagen that form the collagen triple helical molecule. Quantitative impacts on type 1 collagen tend to result in a milder phenotype when compared to qualitative changes due to a dominant-negative effect. Loss-of-function variants generally are associated with classic non-deforming OI with blue sclerae (previously OI type I).
In general, a clear genotype-phenotype correlation does not exist. General rules for genotype-phenotype correlations in COL1A1/2-OI have been published [Ben Amor et al 2011], but there are exceptions to these rules (e.g., glycine to serine substitutions may lead to a more severe phenotype in COL1A1 than a similar change in COL1A2). The extent of variation and the clinical presentation is represented in Maioli et al [2019] (see Figure 2).
Genotype-Phenotype Correlations
It is important to keep the exceptions in mind when providing genetic counseling, particularly in the prenatal setting. Genotyping can be helpful in distinguishing classic non-deforming OI from all other types of OI.
Classic non-deforming OI almost always results from a pathogenic variant in one COL1A1 or COL1A2 allele that introduces premature termination codons and decreases the stability of mRNA (nonsense-mediated decay of the message resulting in a quantitative reduction of the collagen fibril). These causative variants may occur by codon changes, by frame shifts, and by splicing that results in use of cryptic splice sites and premature termination. The type I collagen molecule contains two pro α1(I) chains and a single α2(I) chain. If the number of available pro α1(I) chains decreases, the amount of the trimer manufactured is diminished because no more than one pro α2(I) chain can be accommodated per molecule.
Perinatally lethal OI, progressively deforming OI, and common variable OI all result from pathogenic variants that alter the structure of either pro α1(I) or pro α2(I) chains. This causes a dominant-negative effect whereby the abnormal protein is integrated into the triple helix and collagen fibril, which in turn undergoes continual remodeling, thus resulting in significantly compromised structural integrity of the bone matrix (a qualitative impact on the protein product).
The most common pathogenic variants result in substitution of another amino acid for glycine in the triple helical domain of either chain; serine, arginine, cysteine, and tryptophan result from substitutions in the first position of the glycine codon and alanine, valine, glutamic acid, and aspartic acid result from substitutions in the second position of the glycine codon. Glycine is the least bulky amino acid, and other substituting amino acids do not fit well into the collagen triple helix.
- Substitutions in the pro α1(I) chain by arginine, valine, glutamic acid, aspartic acid, and tryptophan are almost always lethal if they occur in the carboxyl-terminal 70% of the triple helix and have a non-lethal but still moderately severe phenotype if they occur in the remainder of the chain.
- For the smaller side-chain residues (serine, alanine, and cysteine), the phenotypes are more variable and appear to reflect some characteristics of the stability profile of the triple helix that are not yet fully recognized.
- Much more variability occurs with pathogenic variants that affect glycine residues in the pro α2(I) chain, even with the large side-chain residues; therefore, it is more difficult to determine the genotype-phenotype relationship.
The other common disease-causing variants affect splice sites. Variants that lead to exon skipping in the pro α1(I) chain beyond exon 14 and in the pro α2(I) chain beyond exon 25 are generally lethal. The phenotypes resulting from pathogenic variants in the upstream region are more variable and may lead to significant joint hypermobility.
A relatively small number of pathogenic variants that alter amino acid sequences in the carboxyl-terminal regions of both chains have been identified. These domains are used for chain association and pathogenic variants have the capacity to destroy this property or lead to abnormalities in chain association. The phenotypic effects of pathogenic variants that affect this domain appear to be milder when they result in exclusion rather than inclusion of the chain.
Somatic mosaicism for dominant pathogenic variants has been recognized in perinatally lethal OI, progressively deforming OI, and common variable OI. The phenotype of the individual with somatic mosaicism can range from no identifiable characteristics of OI to one of the mild forms. The current estimate for the incidence of somatic/gonadal mosaicism is up to 16% of families.
- Individuals with somatic mosaicism for variants that result in non-lethal forms of OI generally have no phenotypic features of OI, even when the variant is present in a majority of somatic cells.
- Somatic mosaicism for variants that result in lethal OI can produce a mild OI phenotype if the variant is present in the majority of somatic cells; otherwise, the mosaicism is generally asymptomatic.
Penetrance
The penetrance in individuals heterozygous for a COL1A1 or COL1A2 pathogenic variant is 100%, although expression may vary considerably, even in the same family.
Nomenclature
Current (and previously used) nomenclature:
- Classic non-deforming OI with blue sclerae (previously, osteogenesis imperfecta type I)
- Perinatally lethal OI (osteogenesis imperfecta type II)
- Progressively deforming OI (osteogenesis type III)
- Common variable OI with normal sclerae (osteogenesis imperfecta type IV)
The classification scheme of "OI congenita" and "OI tarda" was discarded because fractures at birth can be noted in mild OI and infants with severe OI may not have fractures at birth.
In classifications of genetic conditions, OI may be considered a skeletal dysplasia, a connective tissue disorder, a disorder of collagen or extracellular matrix, or a disorder of bone fragility.
Prevalence
Considering all types, OI has a prevalence of approximately 6-7:100,000. COL1A1/2-OI comprises the largest proportion of OI, representing about 90% of all causes of OI.
Genetically Related (Allelic) Disorders
Other phenotypes associated with germline pathogenic variants in COL1A1 and COL1A2 are summarized in Table 4.
Table 4.
Allelic Disorders
Arterial dissection. Published evidence to date does not support a clear association between spontaneous arterial rupture and COL1A1/2 variants. A study investigating the histopathologic changes in spontaneous carotid artery dissection (sCAD) concluded that no unique connective tissue or vascular phenotype existed in individuals with a known connective tissue disorder when compared to control subjects with ischemic stroke of other etiologies [Dittrich et al 2007].
There have been several isolated case reports of individuals with an identified COL1A1 variant who have suffered spontaneous arterial rupture. Mayer et al [1996] described a G-to-C transversion in one COL1A1 allele resulting in a Gly13Ala substitution in the triple helical domain of the pro α1(I) chain of type I collagen in a woman age 35 years with dissection of the right internal carotid artery and the right vertebral artery after scuba diving. Other than a history of easy bruising and bluish sclerae, she had no evidence of a connective tissue disorder or of OI. Her family history was negative for other individuals with vasculopathy. Malfait et al [2007] describe three unrelated individuals with arginine-to-cysteine substitutions in the pro α1(I) chain who developed iliac or femoral dissection in early adulthood and also had symptoms of classic EDS and osteopenia.
See also Clinical Description, Other Considerations, Cardiovascular for details relating to aortic root dilatation and dissection.
Differential Diagnosis
Distinguishing COL1A1/2 Osteogenesis Imperfecta (COL1A1/2-OI) from Other Types of OI
The primary differential diagnoses for individuals with features of COL1A1/2-OI are non-collagen-associated forms of OI. There are both dominant and recessive types, which can be phenotypically indistinct from COL1A1/2-OI. In a small subset of individuals, specific causative variants have not yet been identified. Table 5 summarizes the molecular basis of these subtypes of OI, the mode of inheritance, the corresponding clinical OI type, and distinguishing clinical and radiographic features.
Table 5.
Other Types of Osteogenesis Imperfecta in the Differential Diagnosis of COL1A1/2 Osteogenesis Imperfecta
Distinguishing OI from Other Disorders and Non-Accidental Trauma
The differential diagnosis of OI depends largely on the age at which the individual is assessed [Plotkin 2004]. Clinical features that help to differentiate COL1A1/2-OI from other conditions include characteristic triangular facies, blue sclerae, joint hypermobility, dental abnormalities, and, in adults, hearing loss.
In Utero Assessment
Early prenatal ultrasound examination or radiographic findings may lead to a consideration of hypophosphatasia, thanatophoric dysplasia, campomelic dysplasia, and achondrogenesis as well as perinatally lethal OI. In some cases, either biochemical or molecular testing can be a useful adjunct.
Table 6.
Genes of Interest in the Differential Diagnosis of COL1A1/2 Osteogenesis Imperfecta – In Utero Assessment
Infancy and Childhood Assessment
Non-accidental trauma (NAT; child abuse). COL1A1/2-OI needs to be distinguished from child physical abuse / non-accidental trauma. The prevalence of physical abuse is much greater than the prevalence of COL1A1/2-OI, and on rare occasion, it can occur in a child with COL1A1/2-OI. Patient history, family history, physical examination, radiographic imaging, and the clinical course all contribute to distinguishing COL1A1/2-OI from child abuse. The overlap in clinical features includes multiple or recurrent fractures, fractures that do not match the history of trauma, and the finding of fractures of varying ages and at different stages of healing [Carty 1988, Ablin et al 1990, Steiner et al 1996, Ablin & Sane 1997, Marlowe et al 2002].
The continued occurrence of fractures in a child who has been removed from a possibly abusive situation lends support to the possibility of COL1A1/2-OI. Metaphyseal and rib fractures, thought to be virtually pathognomonic for child abuse, can rarely occur in COL1A1/2-OI. The presence or absence of blue sclerae is unreliable in distinguishing COL1A1/2-OI from child abuse because blue sclerae are often found in unaffected normal infants until about age 18 months; children with OI type IV may not have blue sclerae.
Family history is often unrevealing; families suspected of possible child abuse often provide an unverified family history of frequent fractures; conversely, the family history of individuals with COL1A1/2-OI often does not reveal any other affected individuals because of a de novo pathogenic variant in the proband or the presence of a mild phenotype in relatives.
Laboratory testing (typically molecular genetic testing of COL1A1 and COL1A2) usually is not needed to differentiate COL1A1/2-OI from NAT, and in some cases, the time required to perform such testing can delay proper disposition of child abuse cases [Steiner et al 1996]. Marlowe and colleagues suggest: "Given the inability to identify all children with OI by clinical examination in situations of suspected non-accidental injury, laboratory testing for OI (and other genetic predispositions for fractures) is a valuable adjunct in discerning the basis for fractures and may identify a small group of children with previously undiagnosed OI" [Marlowe et al 2002]. Laboratory testing in such cases is no substitute for proper clinical evaluation that includes history, family history, physical examination, and radiographic evaluation.
Other genetic disorders. See Table 7.
Table 7.
Other Genetic Disorders of Interest in the Differential Diagnosis of COL1A1/2 Osteogenesis Imperfecta
Idiopathic juvenile osteoporosis (OMIM 259750) typically presents in pre-adolescents with fractures and osteoporosis. The fracture susceptibility and osteoporosis usually resolve spontaneously with puberty.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with COL1A1/2 osteogenesis imperfecta (COL1A1/2-OI), the evaluations summarized in Table 8 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 8.
Recommended Evaluations Following Initial Diagnosis in Individuals with COL1A1/2 Osteogenesis Imperfecta
Treatment of Manifestations
Management focuses on supportive therapy to minimize fractures and maximize function, minimize disability, foster independence, and maintain overall health [Marini & Gerber 1997]. Ideally, COL1A1/2-OI is managed by a multidisciplinary team including specialists in the medical management of COL1A1/2-OI, clinical genetics, orthopedics, rehabilitation medicine, pediatric dentistry, otology/otolaryngology, and mental health.
Supportive therapy is individualized depending on the severity, degree of impairment, and age of the affected individual. Considerable support from medical personnel is generally required by parents caring for infants with perinatally lethal COL1A1/2-OI.
Physical medicine treatment
- Parents and other caregivers should be instructed in safe handling techniques. These are mostly commonsense practices in order to relieve stress on a single point. For example: lift an affected infant by bracing the torso, neck, and lower body; avoid any situation where increased pressure is placed on a single point on any long bone; when assisting an affected child in standing up, do not pull excessively on an extended arm but bend down and brace a greater surface area (e.g., placing a hand behind the back and pulling gently from the front – using the arm – while applying pressure from the rear); avoid sudden acceleration/deceleration movements; and avoid throwing a child in the air. To minimize point pressure, avoid lifting an infant by the ankle when diapering. Older children should not ride on amusement park rides. Caregivers should avoid re-creating the circumstances of a fracture, as it is likely to happen again.
- The use of bracing to try to stabilize progressively deforming limbs depends in part on the subtype of COL1A1/2-OI. Progressively deforming OI has proven to be progressive despite external or internal bracing. The use of internal rods or braces to support and stabilize deforming limbs is more successful in the milder subtypes of COL1A1/2-OI and is guided by the expertise of the managing orthopedist.
- Orthotics to support ankle instability are used in toddlers with delayed walking secondary to joint hypermobility and in other affected individuals who suffer recurrent subluxations of their ankle joints.
- Physical activity serves a number of purposes. It provides gravitational stressors required for bone growth and remodeling. The muscles' supporting joints are strengthened by activity, and as an overall benefit, improved joint stability aids in overall well-being as pain levels are reduced and mobility is increased. Physical activity can be self-directed or coordinated through the services of a physical therapist. Each affected individual's needs are unique and thus both physical and occupational therapy should be initiated for increased stability of bone, improved mobility, prevention of contractures, prevention of head and spinal deformity, and improved aerobic fitness and muscle strengthening.
- Mobility devices, such as scooters and chairs for children and modified automobiles for adults, should be considered.
- Some individuals with COL1A1/2-OI experience chronic daily pain associated with both fractures and nonspecific myofascial pain associated with the generalized connective tissue disorder. Pain management plays an important role in the management of COL1A1/2-OI. Some affected individuals do well with minimal analgesics, but many benefit from a multidisciplinary pain management service. Analgesics can be used to control pain from fractures.
Orthopedic treatment. Fractures are treated as they would be in unaffected children and adults with attention to the following:
- The period of immobility in children with COL1A1/2-OI should be shortened as much as is practical.
- Casts should be small and lightweight.
- Physical therapy should begin as soon as the cast is removed to promote mobility and enhance muscle strength and bone mass.
- At this time, intramedullary rodding remains a mainstay of orthopedic care to provide anatomic positioning of limbs to permit more normal function.
Progressive spinal deformities are particularly difficult to treat because of the poor quality of bone in severely affected children. Progressive scoliosis in severe COL1A1/2-OI may not respond well to conservative management and response to surgical intervention may be limited.
Pharmacologic treatment. Bisphosphonates, analogs of pyrophosphate that decrease bone resorption, are being evaluated in both uncontrolled and controlled trials to assess the extent to which they can increase bone mass and bone strength and improve function in children with COL1A1/2-OI. These studies are still ongoing. Bisphosphonates have been used most extensively in severely affected children; they may be useful in adults as well [Adami et al 2003].
The role of treatment with bisphosphonates in changing the natural history of COL1A1/2-OI is incompletely understood. The Cochrane Collaboration is an international network that assembles reviews on various management strategies based on randomized controlled clinical trials within its database in order to improve the practice of evidence-based medicine. As of the Cochrane Collaboration's most recent update of the OI review, bisphosphonate therapy did not appear to reduce fracture incidence but it did affect bone density and adult height [Dwan et al 2016] (full text).
In a more recent publication Bains et al [2019] collated data from the Osteogenesis Imperfecta Foundation's linked clinical research centers on 466 patients with all forms of OI who had been treated with bisphosphonates. The review of the data primarily focused on classic non-deforming OI with blue sclerae (type I). Primary findings indicated increased lumbar vertebral body density, and statistical regression analysis indicated reduced probability of fracture and scoliosis in individuals treated with bisphosphonates compared with those untreated.
Pamidronate use is invasive and typically requires intravenous infusions every three months, four hours a day, for three days. Pamidronate has been used to treat newborn infants with severe OI; complications include transient asymptomatic hypocalcemia [Plotkin et al 2000] and symptomatic hypocalcemia [Chien et al 2002]. The long-term consequences of lowering bone turnover in children with COL1A1/2-OI are unknown but may include delayed bone union after fracture or osteotomy.
A randomized controlled clinical trial using the oral bisphosphonate alendronate found that treatment with oral alendronate for two years in children with OI significantly decreased bone turnover and increased spine areal BMD (bone mineral content measured by DXA divided by bone area in square centimeters) but was not associated with improved fracture outcomes [Ward et al 2011]. In a second study with a different oral bisphosphonate, Bishop and colleagues found that oral risedronate increased areal BMD and reduced first and recurrent clinical fractures in children with OI [Bishop et al 2010]. Zoledronic acid, a bisphosphonate with a longer half-life, greater potency, and more convenient dosing, has been studied in children with OI. Lv et al [2018] compared the efficacy of an annual infusion of zoledronic acid to weekly oral alendronate and concluded that these treatment approaches had similar increases in vertebral BMD and were well tolerated.
Basilar impression. It is important to screen for this finding so that timely surgical intervention can be planned. A positive "Lhermitte's sign" (tingling in fingers with neck flexion) should prompt neurosurgical referral. Surgery is typically undertaken before persistent/permanent neurologic features are present. If surgery is undertaken, it should be done in a center experienced in the procedures used.
Dental treatment. The goals are the maintenance of both primary and permanent dentition, functional bite or occlusion, optimal gingival health, and overall appearance. Pediatric dentists are the most knowledgeable about dentinogenesis imperfecta (DI) in children. Some consensus exists that early dental restorative coverage of the primary molars and (if possible) aesthetic coverage of the upper anterior teeth is optimal. Plastic polymers are sometimes used to coat teeth. As anxiety can be an issue with children, pre-medication for anxiolysis (e.g., nitrous oxide analgesia or midazolam) can be used for treatment in a clinic setting.
If warranted, orthodontic treatment can be initiated, but care must be taken in the use of orthodontic appliances because of the brittleness of the teeth.
Dental restorations in adults may best be done by a general dentist knowledgeable about OI or a specialist in prosthetic dentistry.
Hearing loss. Surgical repair of the middle-ear bones and creation of a prosthetic incus can improve unaided hearing.
Later hearing loss appears to have a significant sensorineural component that does not respond to middle ear surgery. Cochlear implantation has been used in a small number of individuals; outcome data are limited.
Mental health support through psychiatry/psychology and appropriate social worker intervention can improve quality of life.
Management of lethal OI. It is appropriate to offer parents the option of allowing the infant to expire without attempting heroic interventions such as assisted ventilation.
Other therapies. Early trials of anabolic steroids, sodium fluoride, testosterone, vitamins C and D, flavinoids, and calcitonin showed minimal or no improvement in bone formation, or too small a sample size was utilized for meaningful conclusions [reviewed in Byers & Steiner 1992].
Prevention of Secondary Complications
Special attention should be paid to anesthesia concerns including proper positioning on the operating room table, for which egg crate foam is recommended to avoid fractures.
Surveillance
Table 9.
Recommended Surveillance for Individuals with COL1A1/2 Osteogenesis Imperfecta
Agents/Circumstances to Avoid
Contact sports should be avoided.
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk relatives of an affected individual in order to identify as early as possible those who would benefit from cervical spine examination and dental and hearing evaluations.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Fertility is normal in OI. Pregnancy in women with OI, especially those with progressively deforming OI, can be complicated because of a small pelvis, which may necessitate delivery by cesarean section. For most women who have mild non-deforming OI, pregnancy is uncomplicated. Joint laxity may increase, as it does with unaffected women, and reduce mobility in small, moderately affected women. Bleeding is not more common than usual and complications of vaginal tearing during delivery are not common. Women with OI who are very small require pre-term cesarean section because of respiratory compromise. It is uncertain whether postpartum pelvic relaxation is more common. The mode of delivery of infants with OI has been examined to determine if the frequency of complications is higher with vaginal or cesarean section delivery. No difference in the frequency of complications was found. A higher-than-expected frequency of non-vertex presentations has been noted [Cubert et al 2001]. The role of pregnancy in later fractures, loss of bone mineralization, progression of hearing loss, or any other physical consideration has not been examined in detail.
Women with OI who have significant skeletal deformity and short stature should be followed during pregnancy at a high-risk prenatal care center.
Cesarean section and vaginal delivery of an infant with OI have about the same rate of complications for each type of OI. Delivery of an infant with OI by cesarean section is more frequent than in the general population because a non-vertex presentation cannot be corrected by external manipulation.
Therapies Under Investigation
RANK ligand antibodies, which inhibit osteoclast maturation, have been studied as a therapeutic option in children and results have shown an increase in vertebral body BMD, normalization of vertebral shape, and a reduction in vertebral compression fractures while on therapy. Ongoing studies are determining long-term efficacy and tolerance [Hoyer-Kuhn et al 2014, Hoyer-Kuhn et al 2016, Boyce 2017].
Human growth hormone has been evaluated as an adjunctive therapy in conjunction with bisphosphonates in a randomized controlled study. In this study, growth hormone therapy was reported to correlate with improved linear growth and increased BMD [Antoniazzi et al 2010]. An additional study presented similar results in 26 children with moderate to severe OI when growth hormone was used in isolation [Marini et al 2003].
Teriparatide, a PTH analog, has been used to treat osteoporosis and is being explored as an adjunctive therapy in OI [Orwoll et al 2014]. The risk for osteosarcoma in those treated with teriparatide has limited its widespread use in children with more severe forms of OI. An ongoing registry monitoring the risk in adults treated with teriparatide has not documented an instance of osteosarcoma in the eight years since it was established but longer observation is needed to identify the actual risk of osteosarcoma [Gilsenan et al 2018].
Bone marrow transplantation (BMT) to introduce normal mesenchymal stem cells that have the capacity to differentiate into normal osteoblasts as well as transplanted mesenchymal stromal cells, which produce factors that stimulate endogenous bone growth in individuals with OI, has been evaluated in a pilot clinical trial. Preliminary data suggested a positive impact on growth [Otsuru et al 2012, NCT00187018].
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Classic non-deforming osteogenesis imperfecta (OI), perinatally lethal OI, progressively deforming OI, and common variable OI caused by pathogenic variants in COL1A1 or COL1A2 are inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- Many individuals diagnosed with the milder forms of OI (classic non-deforming and some probands with common variable OI) have an affected parent.
- The proportion of affected individuals who represent simplex cases (i.e., a single occurrence of the disorder in a family) varies by the severity of disease. Approximately 60% of probands with mild OI represent simplex cases. Virtually 100% of probands with progressively deforming or perinatally lethal OI represent simplex cases and have a de novo pathogenic variant or a pathogenic variant inherited from a parent with somatic and/or germline mosaicism.
- Recommendations for the evaluation of parents of a proband with an apparent de novo pathogenic variant include clinical examination of the parents and molecular genetic testing if the variant in the proband has been identified.
- If the causative variant found in the proband cannot be detected in the leukocyte DNA of either parent, possible explanations include a de novo variant in the proband or a very low level of somatic mosaicism that includes the germline in a parent. The overall rate of parental mosaicism is up to 16% in families with dominant COL1A1 or COL1A2 causative variants, although the rate of very low levels of parental mosaicism, which can be difficult to detect in leukocyte DNA using standard molecular genetic techniques, may be higher [Pyott et al 2011, Frederiksen et al 2016].
Sibs of a proband. The risk to the sibs of the proband depends on the clinical/genetic status of the proband's parents:
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs is 50%. Intrafamilial clinical variability is observed among individuals with the same pathogenic variant.
- If the proband has a known COL1A1 or COL1A2 pathogenic or likely pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to the sibs of a proband is about 5% because of the significant possibility of parental somatic and/or germline mosaicism [Pyott et al 2011].
Offspring of a proband. Each child of an individual with a dominantly inherited form of OI has a 50% chance of inheriting the pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected, the parent's family members are at risk.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Considerations in families with an apparent de novo pathogenic variant. When neither parent of a proband with an autosomal dominant condition has the causative variant identified in the proband or clinical evidence of the disorder, the pathogenic variant may be de novo. However, non-medical explanations including alternate paternity or maternity (e.g., with assisted reproduction) and undisclosed adoption could also be explored.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
High-risk pregnancies
- Molecular genetic testing. Once the OI-causing variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
- Biochemical analysis of collagen from fetal cells is no longer offered clinically in the United States.
- Prenatal ultrasound examination performed in a center with experience in diagnosing OI, done at the appropriate gestational age, can be a valuable tool in the prenatal diagnosis of OI. Normally, ultrasound examination detects only the lethal and most severe forms of OI prior to 20 weeks' gestation; milder forms may be detected later in pregnancy when fractures or deformity occurs:
- Perinatally lethal OI. The bony abnormalities can first be seen by ultrasound examination by about 13 to 14 weeks' gestation. By 16 weeks, femoral length is typically two or more weeks delayed, calvarial mineralization is essentially absent, and ribs generally have identified fractures.
- Progressively deforming OI. Limb length generally begins to fall below the growth curve at about 17 to 18 weeks' gestation; serial ultrasound examinations are required to confirm the trend.
Note: Gestational age is expressed as menstrual weeks calculated either from the first day of the last normal menstrual period or by ultrasound measurements.
Low-risk pregnancies. Routine prenatal ultrasound examination may identify a fetus not known to be at risk for COL1A1/2-OI with findings suggestive of OI (perinatally lethal OI or progressively deforming OI) including reduced echogenicity of fetal bones, bowed, crumpled femurs, beaded ribs, evidence of fractures, and markedly diminished calvarial mineralization. As a part of the evaluation of such findings, molecular genetic testing of COL1A1/2 may be considered; however, inability to identify a pathogenic variant does not rule out the diagnosis of OI in the fetus.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- Osteogenesis Imperfecta FoundationPhone: 301-947-0083Fax: 301-947-0456Email: bonelink@oif.org
- Brittle Bone SocietyUnited KingdomPhone: 01382 204446Email: admin@brittlebone.org
- OI RegistryPhone: 844-889-7579Email: bonelink@oif.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
COL1A1/2 Osteogenesis Imperfecta: Genes and Databases
Table B.
OMIM Entries for COL1A1/2 Osteogenesis Imperfecta (View All in OMIM)
Molecular Pathogenesis
Introduction. COL1A1 and COL1A2 encode the α1 and α2 chains of collagen type I, a fibril-forming collagen found in most connective tissues and abundant in bone, cornea, dermis, and tendon. Collagen type I is a heterotrimer consisting of two α1 chains and one α2 chain. It is initially synthesized as a pro α chain with a propeptide at each end (N-propeptide and C-propeptide). The propeptides are necessary for pro α chain association and triple helix formation, which starts at the carboxy-terminal propeptide and extends to the amino-terminal propeptide.
Collagen type I contains a triple helical segment of 1,014 amino acids in which glycine is in every third position and prolines preceding glycine residues are generally hydroxylated, as are some lysyl residues in the Y-position of the Gly-X-Y triplet. Glycine, the smallest amino acid, must be in the third position to allow proper chain folding to occur.
The pathogenic variants in most families are unique; only a few recurrent variants (mostly CpG dinucleotides) are seen in more than one family.
Mechanism of disease causation. In general, the primary mechanism for disease can be viewed as either quantitative or qualitative impacts on collagen type 1 protein. Quantitative changes, which lead to loss of function, tend to have a milder phenotype when compared to qualitative changes, which impart a dominant-negative effect.
Classic non-deforming OI (quantitative, loss of function):
- Decreased production of structurally normal type I procollagen results in a reduction in the amount of bone that can be made, leading to brittle bones.
- The vast majority of disease-causing variants are premature termination codons (e.g., frameshift, nonsense, splice site variants) that result in the reduction of COL1A1 mRNA by half.
Perinatally lethal OI, progressively deforming OI, and common variable OI (qualitative, gain of function):
- Substitutions for glycine within the triple helical domain of the pro α chain delay triple helix formation, resulting in additional post-translation modification that prevents secretion of the assembled trimers.
- Small in-frame deletions or duplications of single amino acids or Gly-X-Y triplets and exon-skipping events may disrupt trimer assembly.
- Diminished amount of type I procollagen is secreted.
- Some of the protein in the matrix has an abnormal structure.
- Clinical consequence is influenced by the position of the substituted glycine, the chain in which the substitution occurs, and the nature of the substituting amino acid.
- Pathogenic variants closer to the 5' end of the protein are likely to result in milder clinical phenotypes due to chain association occurring at the carboxyl-terminal end of the chain.
Chapter Notes
Author Notes
Dr Steiner is a pediatrician, clinical geneticist, and clinical biochemical geneticist. He specializes in general genetics, inherited metabolic diseases, metabolic bone diseases, and osteogenesis imperfecta. Dr Steiner participates in an OI clinic at American Family Children’s Hospital in Madison, Wisconsin for evaluation of children who have or are suspected of having OI.
Dr Basel is the Medical Director for the clinical genetics services for Children’s Wisconsin. He is an Associate Professor at the Medical College of Wisconsin and Chief of the Division of Genetics within the Department of Pediatrics. He is the Associate Director for the Undiagnosed and Rare Disease Program within the Genome Sciences and Precision Medicine Center. He trained under Peter Beighton at the University of Cape Town and worked as a research fellow at University of Connecticut Health Center, studying connective tissue disorders with a focus on fibrillin and collagen 1 disorders.
Author History
Jessica Adsit, MS, CGC; Legacy Center for Maternal Fetal Medicine (2013-2019)
Donald Basel, MD (2013-present)
Peter H Byers, MD; University of Washington Health Sciences Center (2003-2013)
Melanie G Pepin, MS, CGC; University of Washington Health Sciences Center (2003-2013)
Robert D Steiner, MD (2003-present)
Revision History
- 14 March 2024 (aa) Revision: deleted COL1A1 polymorphism NM_00088.4:c.104-441G>T from Genetically Related Disorders (out of scope)
- 6 May 2021 (sw) Revision: updated information on bisphosphonate therapy [Dwan et al 2016]
- 12 December 2019 (aa) Revision: MESD added (Table 5)
- 19 September 2019 (sw) Comprehensive update posted live
- 14 February 2013 (me) Comprehensive update posted live
- 28 January 2005 (me) Review posted live
- 14 June 2003 (rs) Original submission
References
Literature Cited
- Ablin DS, Greenspan A, Reinhart M, Grix A. Differentiation of child abuse from osteogenesis imperfecta. AJR Am J Roentgenol. 1990;154:1035-46. [PubMed: 2108539]
- Ablin DS, Sane SM. Non-accidental injury: confusion with temporary brittle bone disease and mild osteogenesis imperfecta. Pediatr Radiol. 1997;27:111-3. [PubMed: 9028840]
- Adami S, Gatti D, Colapietro F, Fracassi E, Braga V, Rossini M, Tato L. Intravenous neridronate in adults with osteogenesis imperfecta. J Bone Miner Res. 2003;18:126-30. [PubMed: 12510813]
- Antoniazzi F, Monti E, Venturi G, Franceschi R, Doro F, Gatti D, Zamboni G, Tatò L. GH in combination with bisphosphonate treatment in osteogenesis imperfecta. Eur J Endocrinol. 2010;163:479-87. [PubMed: 20592128]
- Ashournia H, Johansen FT, Folkestad L, Diederichsen AC, Brixen K. Heart disease in patients with osteogenesis imperfecta - a systematic review. Int J Cardiol. 2015;196:149-57. [PubMed: 26100571]
- Bains JS, Carter EM, Citron KP, Boskey AL, Shapiro JR, Steiner RD, Smith PA, Bober MB, Hart T, Cuthbertson D, Krischer J, Byers PH, Pepin M, Durigova M, Glorieux FH, Rauch F, Sliepka JM, Sutton VR, Lee B, Nagamani SC, Raggio CL, et al. A multicenter observational cohort study to evaluate the effects of bisphosphonate exposure on bone mineral density and other health outcomes in osteogenesis imperfecta. JBMR Plus. 2019;3:e10118. [PMC free article: PMC6524673] [PubMed: 31131341]
- Balasubramanian M, Verschueren A, Kleevens S, Luyckx I, Perik M, Schirwani S, Mortier G, Morisaki H, Rodrigus I, Van Laer L, Verstraeten A, Loeys B. Aortic aneurysm/dissection and osteogenesis imperfecta: four new families and review of the literature. Bone. 2019;121:191-5. [PubMed: 30684648]
- Basel D, Steiner RD. Osteogenesis imperfecta: recent findings shed new light on this once well-understood condition. Genet Med. 2009;11:375-85. [PubMed: 19533842]
- Ben Amor IM, Glorieux FH, Rauch F. Genotype-phenotype correlations in autosomal dominant osteogenesis imperfecta. J Osteoporos. 2011;2011:540178. [PMC free article: PMC3170785] [PubMed: 21912751]
- Bishop N, Harrison R, Ahmed F, Shaw N, Eastell R, Campbell M, Knowles E, Hill C, Hall C, Chapman S, Sprigg A, Rigby A. A randomized, controlled dose-ranging study of risedronate in children with moderate and severe osteogenesis imperfecta. J Bone Miner Res. 2010;25:32-40. [PubMed: 19580461]
- Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R, Sillence D, Spranger J, Superti-Furga A, Warman M, Unger S. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015;167A:2869-92. [PubMed: 26394607]
- Boyce AM. Denosumab: an emerging therapy in pediatric bone disorders. Curr Osteoporos Rep. 2017;15:283-92. [PMC free article: PMC5554707] [PubMed: 28643220]
- Budsamongkol T, Intarak N, Theerapanon T, Yodsanga S, Porntaveetus T, Shotelersuk V. A novel mutation in COL1A2 leads to osteogenesis imperfecta/Ehlers-Danlos overlap syndrome with brachydactyly. Genes Dis. 2019;6:138-46. [PMC free article: PMC6545454] [PubMed: 31193991]
- Byers PH, Steiner RD. Osteogenesis imperfecta. Annu Rev Med. 1992;43:269-82. [PubMed: 1580589]
- Byers PH, Tsipouras P, Bonadio JF, Starman BJ, Schwartz RC. Perinatal lethal osteogenesis imperfecta (OI type II): a biochemically heterogeneous disorder usually due to new mutations in the genes for type I collagen. Am J Hum Genet. 1988;42:237-48. [PMC free article: PMC1715253] [PubMed: 3341380]
- Carty H. Brittle or battered. Arch Dis Child. 1988;63:350-2. [PMC free article: PMC1778820] [PubMed: 3284477]
- Cepollaro C, Gonnelli S, Pondrelli C, Montagnani A, Martini S, Bruni D, Gennari C. Osteogenesis imperfecta: bone turnover, bone density, and ultrasound parameters. Calcif Tissue Int. 1999;65:129-32. [PubMed: 10430645]
- Charnas LR, Marini JC. Communicating hydrocephalus, basilar invagination, and other neurologic features in osteogenesis imperfecta. Neurology. 1993;43:2603-8. [PubMed: 8255464]
- Chien YH, Chu SY, Hsu CC, Hwu WL. Pamidronate treatment of severe osteogenesis imperfecta in a newborn infant. J Inherit Metab Dis. 2002;25:593-5. [PubMed: 12638943]
- Cole WG, Dalgleish R. Perinatal lethal osteogenesis imperfecta. J Med Genet. 1995.32:284-9. [PMC free article: PMC1050377] [PubMed: 7643358]
- Cole WG, Patterson E, Bonadio J, Campbell PE, Fortune DW. The clinicopathological features of three babies with osteogenesis imperfecta resulting from the substitution of glycine by valine in the pro alpha 1 (I) chain of type I procollagen. J Med Genet. 1992;29:112-8. [PMC free article: PMC1015850] [PubMed: 1613761]
- Cremin B, Goodman H, Spranger J, Beighton P. Wormian bones in osteogenesis imperfecta and other disorders. Skeletal Radiol. 1982;8:35-8. [PubMed: 7079781]
- Cubert R, Cheng EY, Mack S, Pepin MG, Byers PH. Osteogenesis imperfecta: mode of delivery and neonatal outcome. Obstet Gynecol. 2001;97:66-9. [PubMed: 11152910]
- Deodhar AA, Woolf AD. Bone density measurement in osteogenesis imperfecta may well be important. Postgrad Med J. 1994;70:463-4. [PMC free article: PMC2397719] [PubMed: 8029175]
- Dittrich R, Heidbreder A, Rohsbach D, Schmalhorst J, Nassenstein I, Maintz D, Ringelstein EB, Nabavi DG, Kuhlenbäumer G. Connective tissue and vascular phenotype in patients with cervical artery dissection. Neurology. 2007;68:2120-4. [PubMed: 17562832]
- Doi K, Nishimura H, Ohta Y, Kubo T. Stapes surgery in Japanese patients with osteogenesis imperfecta. Adv Otorhinolaryngol 2007;65:226-30. [PubMed: 17245052]
- Dwan K, Phillipi CA, Steiner RD, Basel D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev. 2016;10:CD005088. [PMC free article: PMC6611487] [PubMed: 27760454]
- Emery AEH, Rimoin DL. 2012 Recommendations of the International Committee for Constitutional Disorders of the Skeleton. Elsevier; 2012.
- Faqeih E, Roughley P, Glorieux FH, Rauch F. Osteogenesis imperfecta type III with intracranial hemorrhage and brachydactyly associated with mutations in exon 49 of COL1A2. Am J Med Genet A. 2009;149A:461-5. [PubMed: 19208385]
- Frederiksen AL, Duno M, Johnsen IB, Nielsen MF, Krøigård AB. Asymptomatic parental mosaicism for osteogenesis imperfecta associated with a new splice site mutation in COL1A2. Clin Case Rep. 2016;4:972-8. [PMC free article: PMC5054473] [PubMed: 27761249]
- Gilsenan A, Harding A, Kellier-Steele N, Harris D, Midkiff K, Andrews E. The Forteo Patient Registry linkage to multiple state cancer registries: study design and results from the first 8 years. Osteoporos Int. 2018;29:2335-43. [PMC free article: PMC6154045] [PubMed: 29978254]
- Hayes M, Parker G, Ell J, Sillence D. Basilar impression complicating osteogenesis imperfecta type IV: the clinical and neuroradiological findings in four cases. J Neurol Neurosurg Psychiatry. 1999;66:357-64. [PMC free article: PMC1736265] [PubMed: 10084535]
- Horton WA, Dockery N, Sillence D, Rimoin DL. An embedding method for histochemical studies of undecalcified skeletal growth plate. Stain Technol. 1980;55:19-29. [PubMed: 6158142]
- Hoyer-Kuhn H, Franklin J, Allo G, Kron M, Netzer C, Eysel P, Hero B, Schoenau E, Semler O. Safety and efficacy of denosumab in children with osteogenesis imperfect--a first prospective trial. J Musculoskelet Neuronal Interact. 2016;16:24-32. [PMC free article: PMC5089451] [PubMed: 26944820]
- Hoyer-Kuhn H, Netzer C, Koerber F, Schoenau E, Semler O. Two years' experience with denosumab for children with osteogenesis imperfecta type VI. Orphanet J Rare Dis. 2014;9:145. [PMC free article: PMC4180531] [PubMed: 25257953]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389-97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Kuurila K, Pynnonen S, Grenman R. Stapes surgery in osteogenesis imperfecta in Finland. Ann Otol Rhinol Laryngol. 2004;113:187-93. [PubMed: 15053199]
- Lee JH, Gamble JG, Moore RE, Rinsky LA. Gastrointestinal problems in patients who have type-III osteogenesis imperfecta. J Bone Joint Surg Am. 1995;77:1352-6. [PubMed: 7673285]
- Lund AM, Molgaard C, Muller J, Skovby F. Bone mineral content and collagen defects in osteogenesis imperfecta. Acta Paediatr. 1999;88:1083-8. [PubMed: 10565454]
- Lv F, Liu Y, Xu X, Song Y, Li L, Jiang Y, Wang O, Xia W, Xing X. Zoledronic acid versus alendronate in the treatment of children with osteogenesis imperfecta: a 2-year clinical study. Endocr Pract. 2018;24:179-88. [PubMed: 29466057]
- Maioli M, Gnoli M, Boarini M, Tremosini M, Zambrano A, Pedrini E, Mordenti M, Corsini S, D'Eufemia P, Versacci P, Celli M, Sangiorgi L. Genotype-phenotype correlation study in 364 osteogenesis imperfecta Italian patients. Eur J Hum Genet. 2019;27:1090-100. [PMC free article: PMC6777444] [PubMed: 30886339]
- Malfait F, Symoens S, De Backer J, Hermanns-Lê T, Sakalihasan N, Lapière CM, Coucke P, De Paepe A. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum Mutat. 2007;28:387-95. [PubMed: 17211858]
- Marini JC, Hopkins E, Glorieux FH, Chrousos GP, Reynolds JC, Gundberg CM, Reing CM. Positive linear growth and bone responses to growth hormone treatment in children with types III and IV osteogenesis imperfecta: high predictive value of the carboxyterminal propeptide of type I procollagen. J Bone Miner Res. 2003;18:237-43. [PubMed: 12568401]
- Marini JC, Gerber NL. Osteogenesis imperfecta. Rehabilitation and prospects for gene therapy. JAMA. 1997;277:746-50. [PubMed: 9042848]
- Marlowe A, Pepin MG, Byers PH. Testing for osteogenesis imperfecta in cases of suspected non- accidental injury. J Med Genet. 2002;39:382-6. [PMC free article: PMC1735162] [PubMed: 12070242]
- Mayer SA, Rubin BS, Starman BJ, Byers PH. Spontaneous multivessel cervical artery dissection in a patient with a substitution of alanine for glycine (G13A) in the alpha 1 (I) chain of type I collagen. Neurology. 1996;47:552-6. [PubMed: 8757037]
- Moosa S, Yamamoto GL, Garbes L, Keupp K, Beleza-Meireles A, Moreno CA, Valadares ER, de Sousa SB, Maia S, Saraiva J, Honjo RS, Kim CA, Cabral de Menezes H, Lausch E, Lorini PV, Lamounier A Jr, Carniero TCB, Giunta C, Rohrbach M, Janner M, Semler O, Beleggia F, Li Y, Yigit G, Reintjes N, Altmüller J, Nürnberg P, Cavalcanti DP, Zabel B, Warman ML, Bertola DR, Wollnik B, Netzer C. Autosomal-Recessive Mutations in MESD Cause Osteogenesis Imperfecta. Am J Hum Genet. 2019;105:836-43. [PMC free article: PMC6817720] [PubMed: 31564437]
- Orwoll ES, Shapiro J, Veith S, Wang Y, Lapidus J, Vanek C, Reeder JL, Keaveny TM, Lee DC, Mullins MA, Nagamani SC, Lee B. Evaluation of teriparatide treatment in adults with osteogenesis imperfecta. J Clin Invest. 2014;124:491-8. [PMC free article: PMC3904621] [PubMed: 24463451]
- Otsuru S, Gordon PL, Shimono K, Jethva R, Marino R, Phillips CL, Hofmann TJ, Veronesi E, Dominici M, Iwamoto M, Horwitz EM. Transplanted bone marrow mononuclear cells and MSCs impart clinical benefit to children with osteogenesis imperfecta through different mechanisms Blood. 2012;120:1933-41. [PMC free article: PMC3433095] [PubMed: 22829629]
- Paterson CR, McAllion S, Stellman JL. Osteogenesis imperfecta after the menopause. N Engl J Med. 1984;310:1694-6. [PubMed: 6727948]
- Paterson CR, Mole PA. Bone density in osteogenesis imperfecta may well be normal. Postgrad Med J. 1994;70:104-7. [PMC free article: PMC2397654] [PubMed: 8170878]
- Plotkin H. Syndromes with congenital brittle bones. BMC Pediatr. 2004;4:16. [PMC free article: PMC516444] [PubMed: 15339338]
- Plotkin H, Rauch F, Bishop NJ, Montpetit K, Ruck-Gibis J, Travers R, Glorieux FH. Pamidronate treatment of severe osteogenesis imperfecta in children under 3 years of age. J Clin Endocrinol Metab. 2000;85:1846-50. [PubMed: 10843163]
- Pyott SM, Pepin MG, Schwarze U, Yang K, Smith G, Byers PH. Recurrence of perinatal lethal osteogenesis imperfecta in sibships: parsing the risk between parental mosaicism for dominant mutations and autosomal recessive inheritance. Genet Med. 2011;13:125-30. [PubMed: 21239989]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Robinson ME, Rauch F. Mendelian bone fragility disorders. Bone. 2019;126:11-17. [PubMed: 31039433]
- Sillence DO. Craniocervical abnormalities in osteogenesis imperfecta: genetic and molecular correlation. Pediatr Radiol. 1994;24:427-30. [PubMed: 7700720]
- Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16:101-16. [PMC free article: PMC1012733] [PubMed: 458828]
- Steiner RD, Pepin M, Byers PH. Studies of collagen synthesis and structure in the differentiation of child abuse from osteogenesis imperfecta. J Pediatr. 1996;128:542-7. [PubMed: 8618190]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197-207. [PMC free article: PMC7497289] [PubMed: 32596782]
- van der Rijt AJ, Cremers CW. Stapes surgery in osteogenesis imperfecta: results of a new series. Otol Neurotol 2003;24:717-22. [PubMed: 14501445]
- van Dijk FS, Byers PH, Dalgleish R, Malfait F, Maugeri A, Rohrbach M, Symoens S, Sistermans EA, Pals G. EMQN best practice guidelines for the laboratory diagnosis of osteogenesis imperfecta. Eur J Hum Genet. 2012;20:11-9. [PMC free article: PMC3234509] [PubMed: 21829228]
- van Dijk FS, Huizer M, Kariminejad A, Marcelis CL, Plomp AS, Terhal PA, Meijers-Heijboer H, Weiss MM, van Rijn RR, Cobben JM, Pals G. Complete COL1A1 allele deletions in osteogenesis imperfecta. Genet Med. 2010;12:736-41. [PubMed: 21113976]
- Ward LM, Rauch F, Whyte MP, D'Astous J, Gates PE, Grogan D, Lester EL, McCall RE, Pressly TA, Sanders JO, Smith PA, Steiner RD, Sullivan E, Tyerman G, Smith-Wright DL, Verbruggen N, Heyden N, Lombardi A, Glorieux FH. Alendronate for the treatment of pediatric osteogenesis imperfecta: a randomized placebo-controlled study. J Clin Endocrinol Metab. 2011;96:355-64. [PubMed: 21106710]
- Wenstrup RJ, Willing MC, Starman BJ, Byers PH. Distinct biochemical phenotypes predict clinical severity in nonlethal variants of osteogenesis imperfecta. Am J Hum Genet. 1990;46:975-82. [PMC free article: PMC1683590] [PubMed: 2339695]
Publication Details
Author Information and Affiliations
Medical Director, Newborn Screening, Wisconsin Department of Health Services / Wisconsin State Lab of Hygiene
University of Wisconsin School of Medicine and Public Health
Madison, Wisconsin
Milwaukee, Wisconsin
Publication History
Initial Posting: January 28, 2005; Last Revision: March 14, 2024.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Steiner RD, Basel D. COL1A1/2 Osteogenesis Imperfecta. 2005 Jan 28 [Updated 2024 Mar 14]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.