Summary
Clinical characteristics.
GLI3-related Pallister-Hall syndrome (GLI3-PHS) is characterized by a spectrum of anomalies ranging from polydactyly, asymptomatic bifid epiglottis, and hypothalamic hamartoma at the mild end to laryngotracheal cleft with neonatal lethality at the severe end. Individuals with mild GLI3-PHS may be incorrectly diagnosed as having isolated postaxial polydactyly type A. Individuals with GLI3-PHS can have pituitary insufficiency and may die as neonates from undiagnosed and untreated adrenal insufficiency.
Diagnosis/testing.
The diagnosis of GLI3-PHS can be established in a proband with both hypothalamic hamartoma and mesoaxial polydactyly. Identification of a heterozygous pathogenic variant in GLI3 confirms the diagnosis.
Management.
Treatment of manifestations: Urgent treatment for endocrine abnormalities, especially cortisol deficiency; symptomatic treatment of seizures; elective repair of polydactyly. Management of epiglottic abnormalities depending on the abnormality and the extent of respiratory compromise; bifid epiglottis, the most common abnormality, typically does not need treatment. Treatment of behavioral issues per psychologist &/or psychiatrist; seizures may begin or worsen with use of stimulants for attention-deficit disorder; developmental intervention and/or special education for developmental delays; standard treatment of anal atresia or stenosis.
Surveillance: Annually during childhood: assess growth, monitor for signs of precocious puberty, and assess developmental progress, educational needs, and behavioral issues.
Agents/circumstances to avoid: Biopsy or resection of hypothalamic hamartoma may result in complications and lifelong need for hormone replacement. Some stimulants used for attention-deficit/hyperactivity disorder may exacerbate seizures.
Genetic counseling.
GLI3-PHS is inherited in an autosomal dominant manner. Individuals with GLI3-PHS may have an affected parent or may have the disorder as the result of a de novo pathogenic variant. About 25% of individuals have a de novo pathogenic variant. Persons with a de novo pathogenic variant are generally more severely affected than those with a family history of GLI3-PHS. The risk to offspring of an affected individual is 50%. Prenatal testing for pregnancies at increased risk is possible if the pathogenic variant in the family is known. The reliability of ultrasound examination for prenatal diagnosis is unknown.
Diagnosis
Consensus clinical diagnostic criteria for GLI3-related Pallister-Hall syndrome (GLI3-PHS) have been published [Johnston et al 2010].
Suggestive Findings
GLI3-PHS should be suspected in individuals with the following features:
- Hypothalamic hamartoma, a non-enhancing mass in the floor of the third ventricle posterior to the optic chiasm that is isointense to gray matter on T1- and T2-weighted pulse sequences of an MRI, but may have distinct intensity on FLAIRNote: Neither cranial CT examination nor cranial ultrasound examination is adequate for diagnosis of hypothalamic hamartoma.
- Mesoaxial (i.e., insertional or central) polydactyly, the presence of six or more well-formed digits with a Y-shaped metacarpal or metatarsal
- Postaxial polydactyly (PAP) types A and B. PAP-A is the presence of a well-formed digit on the ulnar or fibular aspect of the limb. PAP-B is the presence of a rudimentary digit or nubbin in the same location.Postaxial polydactyly is probably more common than mesoaxial polydactyly; however, the nonspecificity of postaxial polydactyly and the high frequency of postaxial polydactyly type B in persons of central African descent require caution in its use as a diagnostic feature.
- Bifid epiglottis, a midline anterior-posterior cleft of the epiglottis that involves at least two thirds of the epiglottic leafBifid epiglottis is a useful feature for clinical diagnosis because it appears to be very rare in syndromes other than GLI3-PHS and is also rare as an isolated malformation.
- Other. Imperforate anus, renal abnormalities including cystic malformations, renal hypoplasia, ectopic ureteral implantation, genitourinary anomalies including hydrometrocolpos, pulmonary segmentation anomalies including bilateral bilobed lungs, and nonpolydactyly skeletal anomalies including short limbs
- Family history consistent with autosomal dominant inheritance (e.g., affected males and females in multiple generations). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The clinical diagnosis of GLI3-PHS can be established in a proband with BOTH hypothalamic hamartoma and mesoaxial polydactyly, or the clinico-molecular diagnosis can be established in a proband with suggestive findings and a heterozygous pathogenic (or likely pathogenic) variant in GLI3 identified by molecular genetic testing (see Table 1) [Johnston et al 2010].
Note: (1) Per ACMG/AMP variant classification guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are handled similarly in a diagnostic testing clinical setting, meaning that both can lead to a clinico-molecular diagnosis [Katz et al 2020]. Reference to "pathogenic variants" in this GeneReview is understood to include any pathogenic or likely pathogenic variants. (2) Identification of a heterozygous GLI3 variant of uncertain significance does not establish or rule out the diagnosis. (3) Molecular testing can often identify novel frameshift and nonsense variants in GLI3 in individuals with GLI3-PHS and related phenotypes. Because they are novel, there is often a dearth of clinical and molecular data that can be used to classify the variant. Given that the mechanism of disease does not involve nonsense-mediated decay of the mRNA in these cases, current ACMG/AMP interpretations, with the ClinGen PVS1 criterion specification, may commonly lead to a classification of such a variant as a variant of uncertain significance. It is recommended that an expert with experience in the clinico-molecular diagnosis of this disorder be consulted in this scenario [Lo-Ciganic et al 2019].
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of GLI3-PHS has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of GLI3 to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: To date, large exon or multiexon deletions or duplications have not been reported in individuals with GLI3-PHS.
A multigene panel that includes GLI3 and other genes of interest (see Differential Diagnosis) may be considered to identify the genetic cause of the condition. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in GLI3-Related Pallister-Hall Syndrome
Clinical Characteristics
Clinical Description
GLI3-related Pallister-Hall syndrome (GLI3-PHS) displays a wide range of severity. The literature frequently reflects the assumption that GLI3-PHS is severe and Greig cephalopolysyndactyly syndrome (GCPS) is mild. This assumption is incorrect, as a minority of individuals with GLI3-PHS show multiple severe anomalies and most individuals with GLI3-PHS are mildly affected with polydactyly, asymptomatic bifid epiglottis, and hypothalamic hamartoma. Without careful clinical evaluation, these individuals may be incorrectly diagnosed with postaxial polydactyly type A (PAP-A).
Table 2.
GLI3-Related Pallister-Hall Syndrome: Frequency of Select Features
Hypothalamic hamartoma. Hypothalamic hamartoma is a malformation, not a tumor. Hypothalamic hamartomas grow at the rate of – or more slowly than – the surrounding brain tissue. Hypothalamic hamartomas may be large (≤4 cm in greatest dimension); little correlation exists between the size of the hypothalamic hamartoma and presence or severity of symptoms. Individuals with hypothalamic hamartomas may have neurologic symptoms, although most are asymptomatic. Removal of the hypothalamic hamartoma is not indicated and often results in iatrogenic pituitary insufficiency or other complications.
Endocrine manifestations. The endocrine manifestations of a hypothalamic hamartoma range from isolated growth hormone deficiency or isolated precocious puberty to panhypopituitarism, which can be life threatening. Cortisol deficiency can occur in individuals with nonfamilial GLI3-PHS but appears to be rare in those with familial GLI3-PHS.
Neurologic findings. The best-described neurologic complication of hypothalamic hamartoma is gelastic epilepsy, a partial complex seizure manifest by clonic movements of the chest and diaphragm that simulate laughing. Other types of seizures may be caused by hypothalamic hamartoma. Seizures associated with hypothalamic hamartoma in individuals with GLI3-PHS are generally milder and are responsive to treatment, in contrast to individuals with nonsyndromic hypothalamic hamartoma, who often have refractory seizures [Boudreau et al 2005]. No individual with GLI3-PHS has been shown to have visual field loss even with a hypothalamic hamartoma near the optic chiasm.
Polydactyly. Postaxial polydactyly may be more common than mesoaxial polydactyly in individuals with GLI3-PHS. Postaxial polydactyly (PAP) type A is the presence of a well-formed digit on the ulnar or fibular aspect of the limb. PAP type B is the presence of a rudimentary digit or nubbin in the same location. Mesoaxial (i.e., insertional or central) polydactyly is the presence of six or more well-formed digits with a Y-shaped metacarpal or metatarsal.
Note: The nonspecificity of postaxial polydactyly and the high frequency of PAP type B in persons of central African descent require caution in its use as a diagnostic feature.
Epiglottic abnormalities. Bifid epiglottis is nearly always asymptomatic; however, the more severe clefts of the larynx reported in individuals with GLI3-PHS can cause severe airway symptoms. Posterior laryngeal clefts can be fatal.
Psychiatric and neuropsychological findings. Some individuals with GLI3-PHS have behavioral manifestations, including a few with severe intellectual disability and behavioral disturbances [Ng et al 2004]. A larger study of behavioral manifestations of this disorder was inconclusive, reflecting the difficulty of assessing mild behavioral phenotypes in rare disorders [Azzam et al 2005].
Genitourinary anomalies. Renal abnormalities include cystic malformations, small kidneys, and ectopic ureteral implantation; genitourinary anomalies include hydrometrocolpos. The pathogenetic mechanism of the genitourinary anomalies has been delineated [Blake et al 2016].
Other findings include imperforate anus, pulmonary segmentation anomalies including bilateral bilobed lungs, and nonpolydactyly skeletal anomalies including short limbs.
Prognosis for an individual with GLI3-PHS and no known family history of GLI3-PHS is based on the malformations present in the individual. Literature surveys are not useful for the purpose of establishing the prognosis because there is a large degree of inter-individual heterogeneity and publications tend to show bias of ascertainment to more severe involvement. Although GLI3-PHS has been categorized as a member of the CAVE (cerebroacrovisceral early lethality) group of disorders, few affected individuals have an early lethality phenotype. The CAVE descriptor should be discouraged. Early lethality in GLI3-PHS is most likely attributable to panhypopituitarism that is caused by pituitary or hypothalamic dysplasia or severe airway malformations such as laryngotracheal clefts. In addition, imperforate anus can cause serious complications if not recognized promptly. Thus, in the absence of life-threatening malformations, the prognosis should be assumed to be good for individuals with the nonfamilial occurrence of GLI3-PHS. For individuals with a family history of affected family members, the prognosis is based on the degree of severity present in the family.
Mosaicism. Several individuals with nonsyndromic hypothalamic hamartomas and somatic mosaicism for a GLI3 pathogenic variant in the hamartoma have been reported [Wallace et al 2008]. While these individuals do not meet the clinical diagnostic criteria for GLI3-PHS sensu stricto, they may be considered to have a partial form of GLI3-PHS, and consideration should be given to evaluating such individuals for other manifestations of the disorder.
Sub-PHS. A previous publication suggested the term sub-PHS as a descriptor for those individuals who did not meet the above-specified clinical diagnostic criteria for GLI3-PHS but had some overlapping features and a pathogenic GLI3 variant consistent with the PHS pathogenetic mechanism (see Nomenclature).
Genotype-Phenotype Correlations
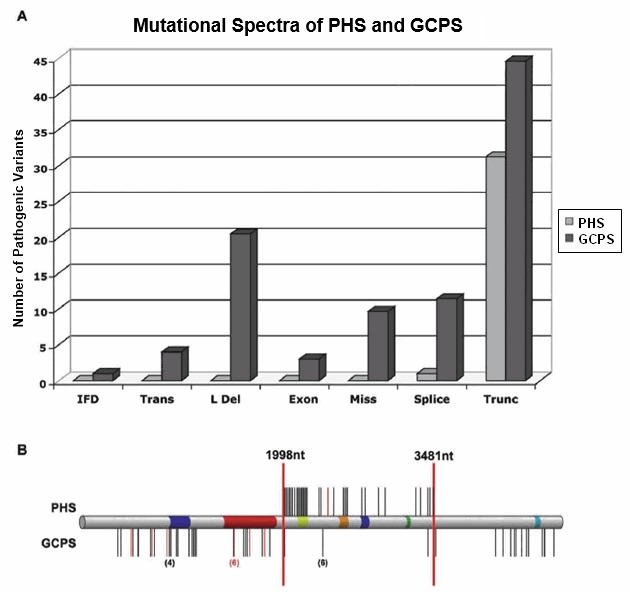
The mutational spectra of GCPS and GLI3-PHS are mostly distinct (see Figure 1). GCPS is caused by pathogenic variants of all types, whereas GLI3-PHS is overwhelmingly caused by truncating variants that generate a frameshift and a truncation. Within the frameshift variant category, a genotype-phenotype correlation has been demonstrated on two levels:
Figure 1.
The mutational spectra of GLI3-PHS and GCPS are mostly distinct.
- Class of variant. Pathogenic variants of all classes can cause GCPS, whereas the majority of pathogenic variants that cause the allelic disorder GLI3-PHS are frameshift variants. Haploinsufficiency for GLI3 causes GCPS, whereas truncating variants 3' of the zinc finger domain of GLI3 generally cause GLI3-PHS [Kang et al 1997] (Figure 1A).
- Variant position. Among all frameshift variants in GLI3, variants in the first third of the gene are only known to cause GCPS (Figure 1B). Frameshift variants in the middle third of the gene cause GLI3-PHS and (uncommonly) GCPS. Frameshift variants in the final third of the gene cause GCPS. No apparent correlation exists between the variant position within each of the three regions and the severity of the corresponding phenotypes.Note: A single truncating variant in the GLI3-PHS region, c.2374C>T (p.Arg792Ter), can cause GCPS and has been observed in six apparently unrelated families [Johnston et al 2005].
Two splice variants have been associated with GLI3-PHS, both presumably causing retention of intron 14, which is also predicted to cause a similarly truncated protein [Al-Qattan et al 2017].
Penetrance
No instances of incomplete penetrance of GLI3-PHS have been published.
Ng et al [2004] reported one individual with apparent germline mosaicism without evident clinical features.
Nomenclature
Other descriptors used include the following:
- Hypothalamic hamartoblastoma syndrome. This is incorrect; "blastoma" refers to tissues in which the neural elements of hamartomas are immature, it does not reflect the syndromic nature of the phenotype, and it may be confused with isolated hamartomas.
- CAVE (cerebroacrovisceral early lethality) complex. This designation is inappropriate as most individuals are mildly affected and do not manifest early lethality.
- Sub-PHS. This designation refers to individuals who do not meet the clinical diagnostic criteria for GLI3-PHS but have some features of PHS and a pathogenic GLI3 variant consistent with the PHS pathogenetic mechanism. However, sub-PHS is probably not a useful term and these individuals should instead be considered to have a mild form of PHS.
- Hall-Pallister syndrome
Note: The abbreviation "HPS" is used for Hermansky-Pudlak syndrome.
Prevalence
GLI3-PHS is rare. The prevalence is unknown. More than 100 affected persons are known to the author [Biesecker, personal observation] and a number of additional individuals have been reported (see, e.g., Démurger et al [2015]). It is suspected that many individuals with postaxial polydactyly and asymptomatic hypothalamic hamartoma or bifid epiglottis may be misdiagnosed as having nonsyndromic PAP-A.
Genetically Related (Allelic) Disorders
Other phenotypes known to be associated with germline pathogenic variants in GLI3 are summarized in Table 3.
Note: Haploinsufficiency for GLI3 causes Greig cephalopolysyndactyly syndrome (GCPS), whereas truncating variants in the middle third of the gene, 3' of the zinc finger domain of GLI3, generally cause GLI3-related Pallister-Hall syndrome (GLI3-PHS) [Johnston et al 2005].
Table 3.
GLI3 Allelic Disorders
Somatic GLI3 pathogenic variants have been identified in individuals with nonsyndromic hypothalamic hamartomas [Wallace et al 2008]. Note: The pathogenicity of a subset of the somatic GLI3 variants reported to be associated with nonsyndromic hypothalamic hamartomas is questioned, as these variants appear to violate the known mechanism of GLI3 pathogenesis.
Differential Diagnosis
Central polydactyly
- Oral-facial-digital syndrome type 6 (OMIM 277170), caused by biallelic pathogenic variants in CPLANE1, includes central polydactyly with hypoplasia of the cerebellar vermis. Renal agenesis and dysplasia have been described.
- Holzgreve syndrome (OMIM 236110) includes central polydactyly, cleft palate, and heart defect.
Postaxial polydactyly. See Table 4.
Table 4.
Disorders Associated with Postaxial Polydactyly in the Differential Diagnosis of GLI3-Related Pallister-Hall Syndrome
Hypothalamic hamartoma. Nonsyndromic or isolated hypothalamic hamartomas may cause either endocrine disturbance (most commonly, growth hormone deficiency or precocious puberty) or a severe neurologic picture of refractory seizures, behavior problems, and cognitive decline. Gelastic epilepsy may be associated [Cohen et al 2021]. Somatic GLI3 pathogenic variants have been identified in nonsyndromic hypothalamic hamartomas (see Genetically Related Disorders) [Wallace et al 2008].
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with GLI3-related Pallister-Hall syndrome (GLI3-PHS), the evaluations summarized in Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 5.
Recommended Evaluations Following Initial Diagnosis in Individuals with GLI3-Related Pallister-Hall Syndrome
Treatment of Manifestations
Table 6.
Treatment of Manifestations in Individuals with GLI3-Related Pallister-Hall Syndrome
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the following evaluations are recommended.
Table 7.
Recommended Surveillance for Individuals with GLI3-Related Pallister-Hall Syndrome
Agents/Circumstances to Avoid
Biopsy or resection of hypothalamic hamartoma may result in complications and lifelong need for hormone replacement.
Some stimulants (commonly used for attention-deficit/hyperactivity disorder) may exacerbate seizures.
Evaluation of Relatives at Risk
Prenatal testing of a fetus at risk. Once the GLI3 pathogenic variant has been identified in an affected family member, prenatal molecular genetic testing may be performed on pregnancies at risk in order to facilitate prompt postnatal treatment of adrenal insufficiency. Adrenal crisis can be lethal in affected infants who have not undergone proper evaluation and treatment for adrenal insufficiency.
Other family members. It is appropriate to evaluate relatives at risk to identify as early as possible those who would benefit from initiation of treatment and preventive measures. Evaluations include:
- Molecular genetic testing for the GLI3 pathogenic variant identified in the proband;
- Clinical examination for polydactyly, laryngoscopy for bifid epiglottis, or MRI for hypothalamic hamartoma. The first-degree relative of a proband is considered affected if hypothalamic hamartoma or central or postaxial polydactyly (PAP) is present in the relative. (PAP type B can be used as a diagnostic criterion for first-degree relatives only in persons who are not of central African descent.)
Note: Assessment for cortisol deficiency must be performed urgently in individuals who have family members with GLI3-PHS and cortisol deficiency.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Pregnancy management of a woman with GLI3-PHS should be attuned to guidelines for the specific manifestations of the disorder. For example, the management of pregnant women with gelastic epilepsy who need to take anticonvulsants is challenging. As there are no guidelines specific to GLI3-PHS, the author recommends following general guidelines for anticonvulsants in pregnancy [Borthen & Gilhus 2012].
In general, women with epilepsy or a seizure disorder from any cause are at greater risk for mortality during pregnancy than pregnant women without a seizure disorder; use of anti-seizure medication (ASM) during pregnancy reduces this risk. However, exposure to ASM may increase the risk for adverse fetal outcome (depending on the drug used, the dose, and the stage of pregnancy at which medication is taken). Nevertheless, the risk of an adverse outcome to the fetus from ASM exposure is often less than that associated with exposure to an untreated maternal seizure disorder. Therefore, use of ASM to treat a maternal seizure disorder during pregnancy is typically recommended. Discussion of the risks and benefits of using a given ASM during pregnancy should ideally take place prior to conception. Transitioning to a lower-risk medication prior to pregnancy may be possible [Sarma et al 2016].
The management of fertility and pregnancy (which is uncommon in individuals with hypopituitarism) in individuals with hypopituitarism caused by GLI3-PHS is similarly challenging and, again, it is recommended that general guidelines be followed [Kübler et al 2009].
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
GLI3-related Pallister-Hall syndrome (GLI3-PHS) is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- Approximately 75% of individuals diagnosed with GLI3-PHS have an affected parent.
- Approximately 25% of individuals diagnosed with GLI3-PHS have the disorder as the result of a de novo GLI3 pathogenic variant. (Individuals with a de novo GLI3 pathogenic variant are generally more severely affected than individuals with a family history of GLI3-PHS [JJ Johnston, unpublished data].)
- If the proband appears to be the only affected family member (i.e., a simplex case), molecular genetic testing is recommended for the parents of the proband to evaluate their genetic status and inform recurrence risk assessment.
- If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.The description of a single parent with germline mosaicism for a GLI3 pathogenic variant [Ng et al 2004] does not allow for estimation of the frequency of this event for genetic recurrence risk estimates, but it must be considered as a possibility.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents.
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%. Because intrafamilial variability appears to be low, affected sibs would be expected to have clinical findings similar to those of the proband.
- If the GLI3 pathogenic variant identified in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental germline mosaicism. One instance of parental mosaicism has been reported [Ng et al 2004].
- If the parents are clinically unaffected but their genetic status is unknown, the risk to the sibs of a proband appears to be low, but greater than that of the general population because of the possibility of parental germline mosaicism. Because there has only been a single occurrence of parental germline mosaicism (and no reports of non-penetrance) reported to date, the parents of a proband with no known family history of GLI3-PHS should be examined for evidence of extra digits. If there is no evidence of extra digits, it is reasonable to conclude that the probability of the parent being heterozygous and nonpenetrant or mosaic is low.
Offspring of a proband. Each child of an individual with GLI3-PHS has a 50% chance of inheriting the GLI3 pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected or has a GLI3 pathogenic variant, the parent's family members may be at risk.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk family members for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the GLI3 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing for GLI3-PHS are possible.
Ultrasound examination. In fetuses at 50% risk, prenatal ultrasound examination may detect polydactyly. However, a normal ultrasound examination does not eliminate the possibility of GLI3-PHS in the fetus.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- American Epilepsy Society
- Epilepsy FoundationPhone: 800-332-1000; 866-748-8008
- Medline Plus
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
GLI3-Related Pallister-Hall Syndrome: Genes and Databases
Table B.
OMIM Entries for GLI3-Related Pallister-Hall Syndrome (View All in OMIM)
Molecular Pathogenesis
GLI3 encodes a zinc finger transcription factor that is downstream of sonic hedgehog in the SHH pathway (SHH-PTCH1-SMO-GLI1, GLI2, GLI3) [Villavicencio et al 2000]. The various GLI proteins in turn transcriptionally regulate genes further downstream in the SHH pathway, including HNF3β, bone morphogenetic proteins, and other as-yet-unknown targets. The human gene is similar to the mouse paralog Gli3 and the vertebrate GLI gene family is homologous to the Drosophila melanogaster gene cubitus interruptus (ci).
Mechanism of disease causation. It has been shown that the truncated forms of the GLI3 protein associated with GLI3-PHS repress transcription [Blake et al 2016]. This explains the relatively specific variant-phenotype association because only a subset of variants can trigger this effect. The variants must truncate the protein without engendering nonsense-mediated decay (which would instead cause the GCPS phenotype).
GLI3-specific laboratory technical considerations. As the result of a cDNA sequencing error, older citations described a longer open reading frame that predicted a protein of 1,596 amino acids; the error has been corrected in the GenBank entry NM_000168.3.
Chapter Notes
Author Notes
The author is a board-certified clinical geneticist and pediatrician. He performs clinical and molecular research on genetic disorders at the National Institutes of Health.
Dr Biesecker is interested in hearing from clinicians treating families affected by GLI3-related PHS who have atypical manifestations or those in whom no causative variant has been identified through molecular genetic testing of the genes known to be involved in this group of disorders. He is also willing to consult on GLI3 variant pathogenicity classifications when the variant or presenting phenotypes are atypical.
Acknowledgments
The author is supported by funding from the Intramural Research Program of the National Human Genome Research Institute.
Revision History
- 22 February 2024 (aa) Revision: information about GLI3 pathogenic variant c.2374C>T (p.Arg792Ter) added to Genotype-Phenotype Correlations
- 18 August 2022 (sw) Comprehensive update posted live
- 18 May 2017 (sw) Comprehensive update posted live
- 18 December 2014 (me) Comprehensive update posted live
- 13 September 2012 (me) Comprehensive update posted live
- 18 March 2008 (me) Comprehensive update posted live
- 6 June 2005 (me) Comprehensive update posted live
- 1 May 2003 (me) Comprehensive update posted live
- 25 May 2000 (me) Review posted live
- 20 January 2000 (lb) Original submission
Note: Pursuant to 17 USC Section 105 of the United States Copyright Act, the GeneReview "GLI3-Related Pallister-Hall Syndrome" is in the public domain in the United States of America.
References
Literature Cited
- Al-Qattan MM, Shamseldin HE, Salih MA, Alkuraya FS. GLI3-related polydactyly: a review. Clin Genet. 2017;92:457-66. [PubMed: 28224613]
- Azzam A, Lerner DM, Peters KF, Wiggs E, Rosenstein DL, Biesecker LG (2005) Psychiatric and neuropsychological characterization of Pallister-Hall syndrome. Clin Genet 67:87-92 [PubMed: 15617553]
- Blake J, Hu D, Cain JE, Rosenblum ND (2016) Urogenital development in Pallister-Hall syndrome is disrupted in a cell-lineage-specific manner by constitutive expression of GLI3 repressor. Hum Mol Genet. 25:437-47. [PMC free article: PMC4731018] [PubMed: 26604140]
- Borthen I, Gilhus NE (2012) Pregnancy complications in patients with epilepsy. Curr Opin Obstet Gynecol. 24:78-83. [PubMed: 22327733]
- Boudreau EA, Liow K, Frattali CM, Wiggs E, Turner JT, Feuillan P, Sato S, Patsalides A, Patronas N, Biesecker LG, Theodore WH (2005) Hypothalamic hamartomas and seizures: distinct natural history of isolated and Pallister-Hall syndrome cases. Epilepsia 46:42-7 [PubMed: 15660767]
- Cohen NT, Cross JH, Arzimanoglou A, Berkovic SF, Kerrigan JF, Miller IP, Webster E, Soeby L, Cukiert A, Hesdorffer DK, Kroner BL, Saper CB, Schulze-Bonhage A, Gaillard WD, et al. Hypothalamic hamartomas: evolving understanding and management. Neurology. 2021;97:864-73. [PMC free article: PMC8610628] [PubMed: 34607926]
- Démurger F, Ichkou A, Mougou-Zerelli S, Le Merrer M, Goudefroye G, Delezoide AL, Quélin C, Manouvrier S, Baujat G, Fradin M, Pasquier L, Megarbané A, Faivre L, Baumann C, Nampoothiri S, Roume J, Isidor B, Lacombe D, Delrue MA, Mercier S, Philip N, Schaefer E, Holder M, Krause A, Laffargue F, Sinico M, Amram D, André G, Liquier A, Rossi M, Amiel J, Giuliano F, Boute O, Dieux-Coeslier A, Jacquemont ML, Afenjar A, Van Maldergem L, Lackmy-Port-Lis M, Vincent-Delorme C, Chauvet ML, Cormier-Daire V, Devisme L, Geneviève D, Munnich A, Viot G, Raoul O, Romana S, Gonzales M, Encha-Razavi F, Odent S, Vekemans M, Attie-Bitach T. New insights into genotype-phenotype correlation for GLI3 mutations.Eur J Hum Genet. 2015;23:92-102. [PMC free article: PMC4266745] [PubMed: 24736735]
- El Mouatani A, Van Winckel G, Zaafrane-Khachnaoui K, Whalen S, Achaiaa A, Kaltenbach S, Superti-Furga A, Vekemans M, Fodstad H, Giuliano F, Attie-Bitach T. Homozygous GLI3 variants observed in three unrelated patients presenting with syndromic polydactyly. Am J Med Genet A. 2021;185:3831-7. [PubMed: 34296525]
- Elson E, Perveen R, Donnai D, Wall S, Black GC. De novo GLI3 mutation in acrocallosal syndrome: broadening the phenotypic spectrum of GLI3 defects and overlap with murine models. J Med Genet. 2002;39:804–6. [PMC free article: PMC1735022] [PubMed: 12414818]
- Everman D. The polydactylies. In: Stevenson RE, Hall JG, eds. Human Malformations and Related Anomalies. 2 ed. Oxford, UK: Oxford University Press; 2006:937-53.
- Johnston JJ, Olivos-Glander I, Killoran C, Elson E, Turner JT, Peters KF, Abbott MH, Aughton DJ, Aylsworth AS, Bamshad MJ, Booth C, Curry CJ, David A, Dinulos MB, Flannery DB, Fox MA, Graham JM, Grange DK, Guttmacher AE, Hannibal MC, Henn W, Hennekam RC, Holmes LB, Hoyme HE, Leppig KA, Lin AE, Macleod P, Manchester DK, Marcelis C, Mazzanti L, McCann E, McDonald MT, Mendelsohn NJ, Moeschler JB, Moghaddam B, Neri G, Newbury-Ecob R, Pagon RA, Phillips JA, Sadler LS, Stoler JM, Tilstra D, Walsh Vockley CM, Zackai EH, Zadeh TM, Brueton L, Black GC, Biesecker LG (2005) Molecular and clinical analyses of Greig cephalopolysyndactyly and Pallister-Hall syndromes: robust phenotype prediction from the type and position of GLI3 mutations. Am J Hum Genet 76:609-22 [PMC free article: PMC1199298] [PubMed: 15739154]
- Johnston JJ, Sapp JC, Turner JT, Amor D, Aftimos S, Aleck KA, Bocian M, Bodurtha JN, Cox GF, Curry CJ, Day R, Donnai D, Field M, Fujiwara I, Gabbett M, Gal M, Graham JM, Hedera P, Hennekam RC, Hersh JH, Hopkin RJ, Kayserili H, Kidd AM, Kimonis V, Lin AE, Lynch SA, Maisenbacher M, Mansour S, McGaughran J, Mehta L, Murphy H, Raygada M, Robin NH, Rope AF, Rosenbaum KN, Schaefer GB, Shealy A, Smith W, Soller M, Sommer A, Stalker HJ, Steiner B, Stephan MJ, Tilstra D, Tomkins S, Trapane P, Tsai AC, Van Allen MI, Vasudevan PC, Zabel B, Zunich J, Black GC, Biesecker LG (2010) Molecular analysis expands the spectrum of phenotypes associated with GLI3 mutations. Hum Mutat. 31:1142-54. [PMC free article: PMC2947617] [PubMed: 20672375]
- Kang S, Graham JM Jr, Olney AH, Biesecker LG. GLI3 frameshift mutations cause autosomal dominant Pallister-Hall syndrome. Nat Genet. 1997;15:266-8. [PubMed: 9054938]
- Katz AE, Nussbaum RL, Solomon BD, Rehm HL, Williams MS, Biesecker LG. Management of secondary genomic findings. Am J Hum Genet. 2020;107:3-14. [PMC free article: PMC7332641] [PubMed: 32619490]
- Kübler K, Klingmüller D, Gembruch U, Merz WM (2009) High-risk pregnancy management in women with hypopituitarism. J Perinatol. 29:89-95. [PubMed: 19177043]
- Lo-Ciganic WH, Donohue JM, Kim JY, Krans EE, Jones BL, Kelley D, James AE, Jarlenski MP. Adherence trajectories of buprenorphine therapy among pregnant women in a large state Medicaid program in the United States. Pharmacoepidemiol Drug Saf. 2019;28:80-9. [PMC free article: PMC6557135] [PubMed: 30192041]
- Ng D, Johnston JJ, Turner JT, Boudreau EA, Wiggs EA, Theodore WH, Biesecker LG (2004) Gonadal mosaicism in severe Pallister-Hall syndrome. Am J Med Genet 124A:296-302 [PubMed: 14708104]
- Rubino S, Qian J, Pinheiro-Neto CD, Kenning TJ, Adamo MA. A familial syndrome of hypothalamic hamartomas, polydactyly, and SMO mutations: a clinical report of 2 cases. J Neurosurg Pediatr. 2018;23:98-103. [PubMed: 30497210]
- Sarma AK, Khandker N, Kurczewski L, Brophy GM. Medical management of epileptic seizures: challenges and solutions. Neuropsychiatr Dis Treat. 2016;12:467-85. [PMC free article: PMC4771397] [PubMed: 26966367]
- Siafa L, Argilli E, Sherr EH, Myers KA. De novo GLI3 pathogenic variants may cause hypotonia and a range of brain malformations without skeletal abnormalities. Pediatr Neurol. 2022;131:1-3. [PMC free article: PMC10257559] [PubMed: 35436645]
- Speksnijder L, Cohen-Overbeek TE, Knapen MF, Lunshof SM, Hoogeboom AJ, van den Ouwenland AM, de Coo IF, Lequin MH, Bolz HJ, Bergmann C, Biesecker LG, Willems PJ, Wessels MW. A de novo GLI3 mutation in a patient with acrocallosal syndrome. Am J Med Genet A. 2013;161A:1394-400. [PubMed: 23633388]
- Villavicencio EH, Walterhouse DO, Iannaccone PM. The sonic hedgehog-patched-gli pathway in human development and disease. Am J Hum Genet. 2000;67:1047-54. [PMC free article: PMC1288546] [PubMed: 11001584]
- Wallace RH, Freeman JL, Shouri MR, Izzillo PA, Rosenfeld JV, Mulley JC, Harvey AS, Berkovic SF (2008) Somatic mutations in GLI3 can cause hypothalamic hamartoma and gelastic seizures. Neurology. 70:653-5. [PubMed: 18057317]
Publication Details
Author Information and Affiliations
National Human Genome Research Institute
National Institutes of Health
Bethesda, Maryland
Publication History
Initial Posting: May 25, 2000; Last Revision: February 22, 2024.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Biesecker LG. GLI3-Related Pallister-Hall Syndrome. 2000 May 25 [Updated 2024 Feb 22]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.