Summary
Clinical characteristics.
RAB18 deficiency is the molecular deficit underlying both Warburg micro syndrome (characterized by eye, nervous system, and endocrine abnormalities) and Martsolf syndrome (characterized by similar – but milder – findings). To date Warburg micro syndrome comprises >96% of reported individuals with genetically defined RAB18 deficiency. The hallmark ophthalmologic findings are bilateral congenital cataracts, usually accompanied by microphthalmia, microcornea (diameter <10), and small atonic pupils. Poor vision despite early cataract surgery likely results from progressive optic atrophy and cortical visual impairment. Individuals with Warburg micro syndrome have severe to profound intellectual disability (ID); those with Martsolf syndrome have mild to moderate ID. Some individuals with RAB18 deficiency also have epilepsy. In Warburg micro syndrome, a progressive ascending spastic paraplegia typically begins with spastic diplegia and contractures during the first year, followed by upper-limb involvement leading to spastic quadriplegia after about age five years, often eventually causing breathing difficulties. In Martsolf syndrome infantile hypotonia is followed primarily by slowly progressive lower-limb spasticity. Hypogonadism – when present – manifests in both syndromes, in males as micropenis and/or cryptorchidism and in females as hypoplastic labia minora, clitoral hypoplasia, and small introitus.
Diagnosis/testing.
The diagnosis of RAB18 deficiency is established in a proband who either has suggestive clinical and neuroimaging findings and biallelic pathogenic variant(s) in RAB3GAP1, RAB3GAP2, RAB18, or TBC1D20 identified by molecular genetic testing or meets the clinical diagnostic criteria when molecular genetic testing has not been performed or has not revealed pathogenic variants in one of the four known genes.
Management.
Treatment of manifestations: Treatment is symptomatic and supportive, and is best approached through collaborative multidisciplinary medical specialists and other professionals. Cataracts are usually removed surgically. Management of developmental delay / intellectual disability and feeding difficulties are as per standard practice. Treatment of seizures is by a neurologist based on seizure type. Motor dysfunction due to progressive spasticity may benefit from physical therapy to maximize mobility and use of durable medical equipment. Undescended testes may require surgical correction; hormone supplementation for hypogonadism may occasionally be undertaken.
Surveillance: Routine follow up with an ophthalmologist, neurologist, developmental specialist, feeding team and nutritionist, and endocrinologist is recommended.
Genetic counseling.
RAB18 deficiency is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the RAB3GAP1, RAB3GAP2, RAB18, or TBC1D20 pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible.
GeneReview Scope
Table
Warburg micro syndrome Martsolf syndrome
Diagnosis
Suggestive Findings
RAB18 deficiency should be suspected in individuals with the following clinical and neuroimaging findings.
Note: Findings indicated with an * are the basis of the diagnosis when molecular genetic testing either has not been performed or has not revealed biallelic pathogenic variants in one of the four known genes.
Clinical Findings
Ophthalmologic
- Bilateral congenital cataracts* (observed in all with a molecularly confirmed diagnosis). Note: To date neither unilateral cataracts nor postnatal cataracts have been observed in persons with molecularly confirmed RAB18 deficiency.
- Bilateral microphthalmia, microcornea* (typically <10 mm in diameter)
- Atonic pupils* (usually constricted and unresponsive to light or mydriatic agents)
- Optic nerve atrophy (usually based on direct inspection of the fundus through a dilated pupil)
- Severe cortical visual impairment. Vision may be light perception only; some may display visual tracking. Electrophysiology confirms cortical visual impairment (see Clinical Description).
Nervous system
- Intellectual disability*. Most affected children do not achieve developmental milestones beyond those of a four-month-old (i.e., they do not achieve independent sitting, crawling, walking, or speech).
- Congenital hypotonia* followed by progressive ascending spasticity associated with contractures of the lower limbs from about 8-12 months; involvement of the upper limbs in later years leads to spastic quadriplegia in most.
- Postnatal microcephaly. Typically in the range of -4 to -6 SD; on occasion may be less pronounced. Congenital microcephaly is rarely observed.
Other
- Short stature
- Hypogonadism. Males: Micropenis and cryptorchidism; females: hypoplastic labia minora, clitoral hypoplasia, and small introitus.
Neuroimaging Findings*
Warburg micro syndrome
- Hypogenesis of the corpus callosum, particularly the splenium.
- Polymicrogyria is bilateral and predominantly frontal, frequently extends to the Sylvian fissure, may extend to the temporal and occipital lobes, and rarely extends over the entire cortex. While polymicrogyria is the most consistently observed cortical abnormality, other described abnormalities include pachygyria and lissencephaly.
- Cortical atrophy: Increased subdural spaces are common.
- Hypoplasia of the cerebellum and cerebellar vermis are common but not universal. Accompanying abnormalities of the pons, as seen in pontocerebellar hypoplasia, are rare.
Martsolf syndrome. Findings are milder than in Warburg micro syndrome, with preserved cortical structure and polymicrogyria usually confined to the frontal lobes.
Establishing the Diagnosis
The diagnosis of RAB18 deficiency is established in a proband who EITHER:
- Has suggestive clinical and neuroimaging findings together with biallelic pathogenic variant(s) in RAB3GAP1, RAB3GAP2, RAB18, or TBC1D20 identified by molecular genetic testing (see Table 1); OR
- Meets the clinical diagnostic criteria outlined under Suggestive Findings when molecular genetic testing has not been performed or when molecular genetic testing has not revealed pathogenic variants in one of the four known genes.
Note that failure to detect biallelic causative pathogenic variant(s) in one of the four genes known to cause RAB18 deficiency does not necessarily exclude a clinical diagnosis of RAB18 deficiency as additional loci may exist.
Molecular testing approaches can include concurrent or serial single-gene testing, use of a multigene panel, and more comprehensive genomic testing.
Gene-targeted testing requires the clinician to determine which gene(s) are likely involved, whereas genomic testing may not. Persons with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom a specific diagnosis has been elusive are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and neuroimaging findings suggest the diagnosis of RAB18 deficiency, molecular genetic testing approaches can include concurrent or serial single-gene testing or use of a multigene panel.
- Serial single-gene testing by sequence analysis can be considered if history of consanguinity together with homozygosity mapping has implicated one of the disease-associated genes as the probable location of a pathogenic variant.Note: Homozygosity mapping describes a specific type of analysis of a SNP chromosomal microarray that identifies regions of homozygosity in affected individuals.
- A multigene panel that includes RAB3GAP1, RAB3GAP2, RAB18, TBC1D20, and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the diagnosis of RAB18 deficiency has not been considered or when gene-targeted testing has not identified pathogenic variant(s), comprehensive genomic testing (typically exome sequencing) is likely to be the diagnostic modality selected.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Molecularly Confirmed RAB18 Deficiency
Clinical Characteristics
Clinical Description
RAB18 deficiency describes the molecular deficit underlying Warburg micro syndrome and Martsolf syndrome. Warburg micro syndrome is characterized by eye, nervous system, and endocrine abnormalities [Warburg et al 1993]; Martsolf syndrome is characterized by similar findings, but with a milder presentation [Martsolf et al 1978]. When first described, Warburg micro syndrome and Martsolf syndrome were considered distinct disorders; however, following discovery of the underlying genetic bases of both phenotypes, it became apparent that these phenotypes are a continuum of clinical manifestations: Warburg micro syndrome on the severe end of the spectrum and Martsolf syndrome at the milder end (see Genotype-Phenotype Correlations).
To date Warburg micro syndrome comprises the majority (>96%) of molecularly confirmed RAB18 deficiency reported [Aligianis et al 2005, Abdel-Salam et al 2007, Yüksel et al 2007, Morris-Rosendahl et al 2010, Bem et al 2011, Borck et al 2011, Dursun et al 2012, Yildirim et al 2012, Handley et al 2013, Liegel et al 2013, Gillespie et al 2014, Picker-Minh et al 2014, Sawyer et al 2014, Imagawa et al 2015, Tasdemir et al 2015, Asahina et al 2016, Gupta et al 2016, Kabzińska et al 2016, Rump et al 2016, Srivastava et al 2016, Trkova et al 2016].
Genetically defined Martsolf syndrome has been described in only four families (8 individuals), whose countries of origin are Pakistan, Mexico, Gambia, and Egypt [Aligianis et al 2006, Handley et al 2013].
Eye Findings
Eye abnormalities are often the first presenting features of RAB18 deficiency. Both Warburg micro syndrome and Martsolf syndrome are associated with the hallmark finding of bilateral congenital cataracts. Cataracts have been observed prenatally by ultrasound examination in several instances and in one instance as early as the second trimester [Morris-Rosendahl et al 2010, Trkova et al 2016].
Cataracts are commonly accompanied by microphthalmia and microcornea (diameter <10 mm). Eyes may appear deep set (enophthalmic). Usually the pupils are small and atonic (i.e., unresponsive to light or mydriatic agents). In rare instances, microphthalmia, microcornea, and atonic pupils are absent in Warburg micro syndrome [Handley et al 2013]. Of the eight individuals reported with Martsolf syndrome, six had microphthalmia; several had small pupils [Aligianis et al 2006, Handley et al 2013].
Despite early cataract surgery, the vision of individuals with RAB18 deficiency remains poor, likely due to progressive optic atrophy and cortical visual impairment, both of which are more severe in Warburg micro syndrome than in Martsolf syndrome. The following electrophysiologic findings support the presence of cortical visual impairment:
- Normal electroretinograms (ERGs)
- Visually evoked potentials (VEPs), which are nearly absent in Warburg micro syndrome but may be present in Martsolf syndrome [Aligianis et al 2005, Aligianis et al 2006, Bem et al 2011, Borck et al 2011, Handley et al 2013, Liegel et al 2013].
Neurologic Findings
Intellectual disability (ID). Individuals with Warburg micro syndrome have severe to profound ID. Individuals with Martsolf syndrome have mild to moderate ID. Language acquisition is largely absent in Warburg micro syndrome. Although acquisition of language is delayed in Martsolf syndrome, several affected children are bilingual [Aligianis et al 2006, Handley et al 2013].
Microcephaly. Postnatal microcephaly is a characteristic – but not invariant – feature of RAB18 deficiency. While the head circumference ranges from -4 to -6 SD in most individuals [Handley et al 2013], it can be within the normal range [Handley et al 2013, Trkova et al 2016]. Conversely, prenatal microcephaly has been seen, on occasion, as a severe manifestation of the disorder [Handley et al 2013].
Feeding difficulties. Individuals with Warburg micro syndrome usually have difficulty feeding and may have gastroesophageal reflux disease and/or dysphagia. Gastrostomy tube placement is usually required to improve nutrition.
Epilepsy. Some – but not all – individuals with RAB18 deficiency have epilepsy. Seizure types vary: focal and generalized seizures, tonic and tonic-clonic seizures, and myoclonic absences have been reported [Graham et al 2004, Handley et al 2013, Mandarano et al 2017]. Within the same family, one sib may have seizures while another sib does not.
EEG findings, which may be normal or abnormal in affected individuals, do not appear to demonstrate a consistent pattern.
Motor Dysfunction
Warburg micro syndrome is associated with progressive ascending spastic paraplegia. Affected infants are usually hypotonic with poor postural and head control; however, they may roll and sit with support. Increased deep tendon reflexes in the lower limbs may progress to hyperreflexia and then contractures and spastic diplegia from approximately 8 to 12 months. Involvement of the upper limbs leading to spastic quadriplegia occurs (variably) from about age five years. Later progression may lead to breathing difficulties. It is likely that paraplegia results from impaired motor neuron function.
Clinical findings of a motor and sensory peripheral neuropathy have been reported [Nassogne et al 2000, Graham et al 2004, Kabzińska et al 2016].
Martsolf syndrome is associated with hypotonia and primarily lower-limb spasticity, which progresses more slowly than that of Warburg micro syndrome [Aligianis et al 2006, Handley et al 2013]. Three of the eight individuals reported with Martsolf syndrome were able to walk with assistance.
Hypogonadism
Both Warburg micro syndrome and Martsolf syndrome are frequently associated with hypogonadism. Clinical findings are consistent with hypogonadotropic hypogonadism of hypothalamic origin [Warburg et al 1993, Graham et al 2004, Handley et al 2013, Asahina et al 2016].
In affected males it manifests as micropenis and/or cryptorchidism [Bem et al 2011, Handley et al 2013]. In affected females, it may manifest as hypoplastic labia minora, clitoral hypoplasia, and small introitus [Graham et al 2004, Handley et al 2013].
Untreated hypogonadism may be associated with delayed-onset puberty or lack of puberty.
Other
RAB18 deficiency is often – but not always – associated with short stature (height <-2 SD).
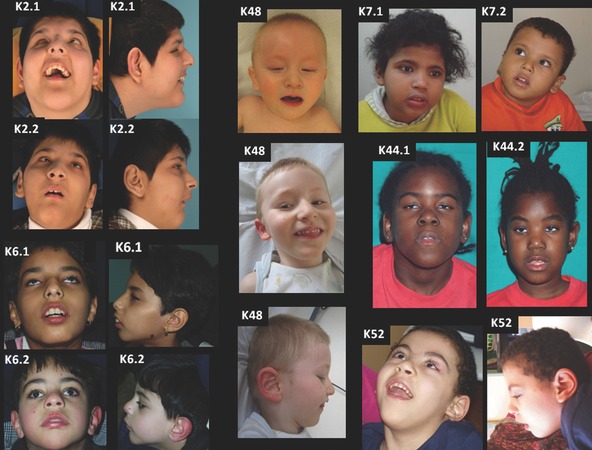
Dysmorphic features associated with RAB18 deficiency are mild but form a recognizable pattern, including deep-set eyes, a wide nasal bridge and prominent nasal root, relatively narrow mouth, and proportionately large anteverted ears (Figure 1).

Figure 1.
Photographs of individuals with Warburg micro and Martsolf syndromes Brothers K2.1 (age 15 years) and K2.2 (age 13 years) with Warburg micro syndrome are homozygous for the RAB3GAP1 p.Thr18Pro variant.
Mild micrognathia, a high-arched palate, and delayed dentition may also be seen [Handley et al 2013, Aligianis & Handley 2016].
Mild hypertrichosis has been reported; see for example Yüksel et al [2007], Morris-Rosendahl et al [2010], Picker-Minh et al [2014], and Mandarano et al [2017].
Osteopenia has been reported as potentially a primary manifestation of Warburg micro syndrome in one family [Picker-Minh et al 2014].
Although secondary complications from RAB18 deficiency can be life-threatening, RAB18 deficiency is not known to directly affect life expectancy.
Phenotype Correlations by Gene
Clinical observations cannot be used to distinguish the genetic basis of RAB18 deficiency.
The strongest suggestion of phenotype correlation by gene comes from comparative analysis of brain MRIs in which it appears that biallelic loss-of-function RAB3GAP2 variants may result in milder malformations than biallelic variants in either RAB3GAP1 or RAB18 [Handley et al 2013].
Of note, several individuals with biallelic loss-of-function pathogenic variants in TBC1D20 have developed glaucoma [Liegel et al 2013], an uncommon finding in RAB18 deficiency caused by biallelic variants in the other three genes (RAB3GAP1, RAB3GAP2, or RAB18).
Genotype-Phenotype Correlations
RAB18 deficiency results from biallelic pathogenic variants in RAB3GAP1, RAB3GAP2, RAB18, or TBC1D20 [Aligianis et al 2005, Aligianis et al 2006, Bem et al 2011, Borck et al 2011, Liegel et al 2013]. Biallelic loss-of-function variants in any one of these genes cause Warburg micro syndrome, which corresponds to the severe end of the phenotypic spectrum. Pathogenic variants that diminish but do not completely nullify gene expression or function can cause or contribute to Martsolf syndrome, which corresponds to the milder end of the phenotypic spectrum.
Warburg micro syndrome comprises the majority (98/102 families) of molecularly confirmed RAB18 deficiency reported [Aligianis et al 2005, Abdel-Salam et al 2007, Yüksel et al 2007, Morris-Rosendahl et al 2010, Bem et al 2011, Borck et al 2011, Dursun et al 2012, Yildirim et al 2012, Handley et al 2013, Liegel et al 2013, Gillespie et al 2014, Picker-Minh et al 2014, Sawyer et al 2014, Tasdemir et al 2015, Asahina et al 2016, Gupta et al 2016, Imagawa et al 2015, Kabzińska et al 2016, Rump et al 2016, Srivastava et al 2016, Trkova et al 2016].
- RAB18 deficiency usually results from pathogenic variants likely to completely nullify gene expression (nonsense, frameshift, intragenic deletion or consensus splice sites). The clinical presentation in these individuals is highly consistent, in keeping with the likelihood that the clinical consequences of these loss-of-function variants are equivalent.
- Other variants associated with Warburg micro syndrome that are likely to abrogate the function of the encoded protein (see Molecular Genetics) include:
- RAB3GAP1: Three missense variants [Handley et al 2013, Asahina et al 2016].
- RAB18: Two missense variants, an in-frame deletion, and an extension variant [Bem et al 2011, Handley et al 2013].
Martsolf syndrome. The three pathogenic variants reported in four families with Martsolf syndrome [Aligianis et al 2006, Handley et al 2013] are as follows:
- RAB3GAP1. A homozygous frameshift variant, c.9delC, identified in affected sibs, inactivates the normal RAB3GAP1 transcript but also expresses a novel transcript whose protein product may provide sufficient residual function to result in the milder phenotype of Martsolf syndrome [Handley et al 2013] (see details in Molecular Genetics).
- RAB3GAP2
- The homozygous missense variant c.3154G>T (Gly1052Cys) was identified in three affected individuals from one family [Aligianis et al 2006]. In lymphocyte RNA from two of the affected individuals, this variant was found to promote exon-skipping and to introduce a frameshift into the shortened transcript; however, some full-length transcript was also found in each case, compatible with residual full-length protein expression.
- The homozygous missense variant c.1276C>T (p.Arg426Cys) affects a conserved amino acid residue, and the altered protein may retain some functional activity [Handley et al 2013].
Nomenclature
Although the following naming system has been used – in some instances – to designate the causative gene for Warburg micro syndrome, no clinical purpose is served by this naming system as the causative gene does not influence phenotype.
- Warburg micro syndrome type 1: RAB3GAP1
- Warburg micro syndrome type 2: RAB3GAP2
- Warburg micro syndrome type 3: RAB18
- Warburg micro syndrome type 4: TBC1D20
Prevalence
Data on the prevalence of RAB18 deficiency are limited.
As expected for an autosomal recessive disorder, incidence is known to be higher in isolated communities and in communities with a high rate of consanguinity [Bem et al 2011, Handley et al 2013].
Incidence may be higher in populations in which pathogenic founder variants are present at a high frequency. Notably, loss of function of an essential splice site variant in RAB3GAP1, c.748+1G>A, has been identified as a founder allele in 11 families of Turkish origin [Aligianis et al 2005, Yüksel et al 2007, Dursun et al 2012, Yildirim et al 2012, Handley et al 2013, Tasdemir et al 2015].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in RAB3GAP1, RAB3GAP2, RAB18, or TBC1D20.
Differential Diagnosis
Array CGH may be useful in identifying pathogenic copy number variants associated with clinical findings similar to those in RAB18 deficiency [Arroyo-Carrera et al 2015]. Similarly, TORCH screening may useful in determining whether prenatal infection is a potential cause of the clinical findings [Gupta et al 2016].
Note that in RAB18 deficiency, prenatal onset of cataracts appears to be a consistent feature, a finding in contrast to other inherited conditions in which cataracts are a more variable manifestation or can arise postnatally (Table 2).
Differential diagnosis therefore includes some other syndromes with congenital cataracts.
Table 2.
Disorders to Consider in the Differential Diagnosis of RAB18 Deficiency
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with RAB18 deficiency, the evaluations in Table 3 (if not performed as part of the evaluation that led to diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis of RAB18 Deficiency
Treatment of Manifestations
Treatment is symptomatic and supportive. It is best approached through collaborative care involving medical specialists, and is best coordinated by a pediatrician who is aware of the child's general health and development. Psychosocial support for families is an important component of effective care.
Table 4.
Treatment of Manifestations in Individuals with RAB18 Deficiency
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, and feeding therapy. In the US, early intervention is a federally funded program available in all states.
Ages 3-5 years. Patient is evaluated to determine required services and therapies, and to develop an individualized education plan (IEP).
Ages 5-21 years. Developmental pediatricians can provide recommendations for schooling and assistance with transition to adulthood.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate agencies and to support parents in maximizing quality of life.
Consideration of private supportive therapies based on the affected individual's needs is recommended. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
In the US:
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC] for individuals who have expressive language difficulties.
Prevention of Secondary Complications
Respiratory infections. Individuals with Warburg micro syndrome are prone to respiratory infection because of reduced mobility, aspiration, and motor dysfunction. These can be life threatening and should be treated accordingly.
Risk of injury. Reduced mobility, hormonal imbalance, and treatment with anticonvulsants are risk factors for the development of osteoporosis. Affected individuals are at increased risk of broken limbs and dislocations and may be unable to effectively communicate the cause of the resulting distress. Consideration should be given to ongoing monitoring of bone densities and vitamin D levels, and to orthopedic assessment in case of acute admissions.
Surveillance
No formal surveillance guidelines exist for RAB18 deficiency. Suggested surveillance includes routine follow up with
- An ophthalmologist
- A neurologist
- A developmental specialist, such as a developmental pediatrician
- A feeding team and nutritionist
- An endocrinologist, especially in infancy to assess degree of hypogonadism and at the age that puberty typically would occur
- Hearing tests
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
RAB18 deficiency is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., carriers of one RAB3GAP1, RAB3GAP2, RAB18, or TBC1D20 pathogenic variant).
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a RAB3GAP1, RAB3GAP2, RAB18, or TBC1D20 pathogenic variant.
Carrier (Heterozygote) Detection
Carrier testing for at-risk relatives requires prior identification of the RAB3GAP1, RAB3GAP2, RAB18, or TBC1D20 pathogenic variants in the family.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are carriers or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown).
Prenatal Testing and Preimplantation Genetic Testing
Once the RAB3GAP1, RAB3GAP2, RAB18, or TBC1D20 pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for RAB18 deficiency are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
No specific resources for RAB18 Deficiency have been identified by GeneReviews staff.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
RAB18 Deficiency: Genes and Databases
Table B.
OMIM Entries for RAB18 Deficiency (View All in OMIM)
Molecular Pathogenesis
The molecular pathology of Warburg micro syndrome and Martsolf syndrome arises either from the absence of Ras-related protein Rab-18 (RAB18) protein or from its functional absence as a result of dysregulation. Because RAB3GAP1, RAB3GAP2, and TBC1D20 are each essential for the regulation of RAB18, biallelic loss-of-function variants in the genes that encode these proteins lead to clinically indistinguishable phenotypes.
RAB18 encodes a highly conserved member of the RAB subfamily of the RAS superfamily of small GTPases [Handley 2017]. Different RAB protein isoforms function to regulate discrete steps in trafficking between cellular membrane compartments. RAB18 is proposed to have roles in regulation of lipid droplets, lipolysis, and lipogenesis [Martin et al 2005, Ozeki et al 2005, Pulido et al 2011], trafficking between the Golgi and endoplasmic reticulum (ER) [Dejgaard et al 2008, Handley et al 2015], ER structure [Gerondopoulos et al 2014], exocytosis [Vazquez-Martinez et al 2007], and autophagy [Feldmann et al 2017]. The specific cellular deficit(s) that underlie the pathology of Warburg micro syndrome and Martsolf syndrome are not yet known.
In common with other small GTPases, RAB proteins function as "molecular switches." They can bind to GDP or GTP and adopt different conformations according to which nucleotide is bound. These different conformations are in turn associated with altered protein-binding characteristics that affect interactions with regulators and with the mediators of their downstream cellular functions. Switching between GDP- and GTP-bound conformations is tightly regulated by two classes of protein, guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). GEFs mediate the exchange of bound GDP for GTP. GAPs stimulate the intrinsic GTPase activity of the RAB proteins, thereby mediating hydrolysis of bound GTP into GDP.
RAB3GAP1 and RAB3GAP2 each encode essential subunits of a binary complex with GEF activity toward RAB18 [Gerondopoulos et al 2014]. This complex is necessary to mediate GDP-GTP exchange and the associated RAB18 conformational change.
TBC1D20 encodes a RAB-GAP with modest in vitro GAP activity toward RAB18. Several lines of evidence indicate that it functions as a RAB18-GAP physiologically [Haas et al 2007, Handley et al 2015]. The regulation of RAB18 by TBC1D20 opposes that of the RAB3GAP complex, promoting its GDP- rather than the GTP-bound conformation. However, both regulators are required for RAB18 to function appropriately, in a spatiotemporally restricted manner.
RAB3GAP1
Gene structure. The canonic RAB3GAP1 transcript, NM_012233.2, comprises 24 coding exons and encodes a 981-amino acid protein.
An additional in-frame microexon of 21 nucleotides constitutes exon 24 that is incorporated into the 25-exon alternative transcript, NM_001172435; this microexon may be differentially expressed in the brain [Irimia et al 2014]. For a detailed summary of gene and protein information, see Table A, Gene; a summary of transcripts is in Ensemble.
Pathogenic variants. Biallelic pathogenic variants in RAB3GAP1 are the most frequently reported cause of RAB18 deficiency. Pathogenic variants are found throughout the gene and in most cases are likely to compromise gene expression.
- Homozygous pathogenic variants were identified in 67 families: 19 were splice site, 18 nonsense, 20 frameshift, six missense, and four intragenic deletions [Aligianis et al 2005, Abdel-Salam et al 2007, Yüksel et al 2007, Morris-Rosendahl et al 2010, Dursun et al 2012, Yildirim et al 2012, Handley et al 2013, Gillespie et al 2014, Picker-Minh et al 2014, Sawyer et al 2014, Imagawa et al 2015, Tasdemir et al 2015, Gupta et al 2016, Kabzińska et al 2016, Rump et al 2016, Srivastava et al 2016, Patel et al 2017].
- Compound heterozygous pathogenic variants were identified in ten families: 12 were nonsense, three splice site, three frameshift, one missense, and one was unidentified [Handley et al 2013, Asahina et al 2016, Trkova et al 2016].
Pathogenic variants identified in RAB3GAP1 in more than one family include:
- In individuals of Turkish ethnic origin: the splice site variant c.748+1G>A [Aligianis et al 2005, Yüksel et al 2007, Dursun et al 2012, Yildirim et al 2012, Handley et al 2013, Tasdemir et al 2015].
- In individuals of Pakistani ethnic origin: the splice site variant c.649-2A>G and a missense variant, c.52A>C [Aligianis et al 2005, Handley et al 2013].
Recurrent pathogenic variants identified in individuals with different ethnic backgrounds include c.899+1G>A, c.1039C>T, and c.2801delC.
The apparent frameshift variant c.9delC was identified in sibs with Martsolf syndrome [Handley et al 2013] (see Genotype-Phenotype Correlations). Analysis of lymphoblast RNA from an affected child detected expression of an alternate RAB3GAP1 transcript, which may be sufficient to explain the milder Martsolf syndrome phenotype in this family.
Table 5.
RAB3GAP1 Pathogenic Variants Discussed in This GeneReview
Normal gene product. Functional characterization of RAB3GAP1 protein domains is limited. Biochemical evidence suggests that GTPase-activating protein ("GAP") activity specific for RAB3 isoforms resides in the C-terminal portion of the protein between amino acid residues 600 and 981 and identifies Arg728 as a critical residue in this activity [Clabecq et al 2000]. No evidence to date has definitively implicated loss of RAB3-GAP activity in, or excluded it from, a role in disease pathogenesis.
Abnormal gene product. Biochemical characterization of several of the disease-associated variants in RAB3GAP1 has been carried out [Gerondopoulos et al 2014].
Two of three pathogenic missense variants in RAB3GAP1, resulting in p.Thr18Pro and p.Glu24Val substitutions, disrupt the in vitro RAB18-GEF activity of the RAB3GAP1-RAB3GAP2 complex but do not affect the in vitro RAB3-GAP activity of RAB3GAP1. These data suggest that loss of RAB18-GEF activity is responsible for disease pathogenesis.
A third pathogenic missense variant, c.560G>C, p.Arg187Pro, has not been characterized at a molecular level [Asahina et al 2016].
The location of the loss-of-function missense variants in RAB3GAP1 may indicate that the N-terminal region of RAB3GAP1 is critical for its RAB18-GEF activity. However, the association of a c.9delC variant with Martsolf syndrome rather than Warburg micro syndrome [Handley et al 2013] suggests that the extreme N-terminus may be dispensable for the protein to retain some functional activity.
Two of the pathogenic variants in RAB3GAP1, c.2801delC and c.2865_2866insTTCT, affect the last exon of the gene and are therefore unlikely to reduce protein expression as a result of nonsense-mediated decay of the transcript [Aligianis et al 2005, Handley et al 2013]. This suggests that residues at the C-terminus of the protein, amino acids 934-981, are functionally important.
RAB3GAP2
Gene structure. The canonic RAB3GAP2 transcript, NM_012414, comprises 35 coding exons and encodes a 1,393-amino acid protein. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. In all affected individuals in the 11 families reported to date with RAB3GAP2 pathogenic variants, the variants have been homozygous [Aligianis et al 2006, Borck et al 2011, Handley et al 2013].
Nonsense variants were identified in five families, frameshift variants in two families, and missense variants in two families. An in-frame deletion and a variant affecting splicing were each identified in single families. One pathogenic missense RAB3GAP2 variant, c.1276C>T, p.Arg426Cys, was associated with Martsolf syndrome in two families of different ethnic origins.
Table 6.
RAB3GAP2 Pathogenic Variants Discussed in This GeneReview
Normal gene product. Functional characterization of RAB3GAP2 protein domains is limited.
Abnormal gene product. Characterization of the pathogenic p.Arg426Cys variant in RAB3GAP2 shows that this substitution disrupts the in vitro RAB18-GEF activity of the RAB3GAP1-RAB3GAP2 complex [Gerondopoulos et al 2014].
An in-frame deletion associated with Warburg micro syndrome, c.499_507del, removes conserved residues Phe167, Tyr168, and Thr169 from the protein, suggesting that these residues are also functionally important [Borck et al 2011].
RAB18
Gene structure. The canonic RAB18 transcript, NM_021252.4, comprises seven coding exons and encodes a 206-amino acid protein. Multiple poorly characterized alternatively-spliced RAB18 transcripts have been identified. One such transcript, NM_001256410, incorporates an additional exon encoding 29 additional amino acids, but this transcript has not been associated with disease. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. In nine families in which pathogenic variants have been reported to date, the variants were homozygous in eight and compound heterozygous in one [Bem et al 2011, Handley et al 2013, Gillespie et al 2014, Mandarano et al 2017].
A pathogenic founder variant, c.71T>A, is responsible for RAB18 deficiency in five families of ethnic Pakistani origin [Bem et al 2011, Gillespie et al 2014]. Other identified pathogenic variants are in Table 7.
Table 7.
RAB18 Pathogenic Variants Discussed in This GeneReview
Normal gene product. A crystal structure of RAB18 has been generated (Protein Data Bank reference: 1X3S). In this structure, residues Gly15-Ser23, Phe33-Thr40, Asp63-Gln67, Asn122-Lys126, and Ala150-Lys153 are each in close proximity to bound nucleotide. Residues Lys21-Ser22, Ala38-Thr40, and Asp63-Thr64 are each in close proximity to bound magnesium. RAB18 has a C-terminal Cys-Ser-Val-Leu motif (amino acids 203-206) that is subject to post-translational modification. The Cys residue is geranylgeranylated, the terminal Ser-Val-Leu is then cleaved and the Cys residue is methylated [Leung et al 2007]. The geranylgeranyl-modification is important for the interactions of RAB18 with regulatory molecules and with membranes.
Abnormal gene product. A comparable clinical picture in individuals with a nonsense variant, a deletion of exon 2, and other pathogenic variants in this gene suggests that each of these variants completely abrogates protein function [Bem et al 2011, Mandarano et al 2017].
In vitro analyses of recombinant RAB18 p.Leu24Gln and p.Arg93del proteins shows that these variants completely abolish nucleotide binding [Bem et al 2011].
A c.284C>G, p.Thr95Arg variant has not been functionally characterized, but the proximity of Thr95 to Arg93 suggests that this substitution has a similar effect [Handley et al 2013].
A disease-associated extension variant, c.619T>C, is unlikely to impair nucleotide binding but is likely to compromise the post-translational modification of RAB18 and its association with membranes [Bem et al 2011].
Together, these data suggest that both nucleotide binding and post-translational modification are essential for RAB18 to function.
TBC1D20
Gene structure. The canonic TBC1D20 transcript, NM_144628.3, comprises eight coding exons and encodes a 403-amino acid protein (NP_653229.1).
Pathogenic variants. Pathogenic TBC1D20 variants have been homozygous in the affected children in all five families reported to date [Liegel et al 2013]. Nonsense variants were identified in three families; a frameshift variant and a deletion encompassing exons 2-8 were each identified in single families.
Normal gene product. TBC1D20 is a member of a family of homologous proteins, each containing a Tre2-Bub2-Cdc16 (TBC) domain. TBC domains have been found to confer RAB-GAP activity on these proteins, and each usually has a differing specificity for different RAB protein isoforms [Frasa et al 2012]. The TBC domain of TBC1D20 is located between amino acids 60 and 246. A crystal structure of TBC1D20 in a complex with RAB1B (Protein Data Bank reference: 4HLQ) has been described, and amino acid residues Arg105 and Gln144 shown to be important for catalytic activity [Gavriljuk et al 2012]. TBC1D20 contains a C-terminal transmembrane domain between amino acids 367 and 387 through which it associates with the membrane of the endoplasmic reticulum; this domain is likely to be essential for normal protein function.
Abnormal gene product. Each of the identified human pathogenic variants in TBC1D20 is likely to disrupt protein expression. A comparable clinical picture between an individual with a deletion encompassing exons 2-8 and the other affected individuals suggests that loss of protein expression is complete in each instance [Liegel et al 2013].
A pathogenic variant identified in the mutated mouse model, blind-sterile, is an in-frame deletion affecting five amino acids corresponding to Phe232-Val236 in the TBC domain of the human protein. The mutant protein of blind-sterile shows reduced stability and dramatically reduced catalytic activity as compared to its wild type counterpart suggesting that these residues are functionally important [Liegel et al 2013].
References
Literature Cited
- Abdel-Salam GM, Hassan NA, Kayed HF, Aligianis IA. Phenotypic variability in Micro syndrome: report of new cases. Genet Couns. 2007;18:423–35. [PubMed: 18286824]
- Aligianis IA, Handley MT. RAB3GAP1, RAB3GAP2, RAB18, TBC1D20, and the Warburg micro and Martsolf syndromes. In: Erickson RP, Wynshaw-Boris AJ, eds. Epstein's Inborn Errors of Development. 3 ed. New York, NY: Oxford University Press. 2016.
- Aligianis IA, Johnson CA, Gissen P, Chen D, Hampshire D, Hoffmann K, Maina EN, Morgan NV, Tee L, Morton J, Ainsworth JR, Horn D, Rosser E, Cole TR, Stolte-Dijkstra I, Fieggen K, Clayton-Smith J, Megarbane A, Shield JP, Newbury-Ecob R, Dobyns WB, Graham JM Jr, Kjaer KW, Warburg M, Bond J, Trembath RC, Harris LW, Takai Y, Mundlos S, Tannahill D, Woods CG, Maher ER. Mutations of the catalytic subunit of RAB3GAP cause Warburg micro syndrome. Nat Genet. 2005;37:221–3. [PubMed: 15696165]
- Aligianis IA, Morgan NV, Mione M, Johnson CA, Rosser E, Hennekam RC, Adams G, Trembath RC, Pilz DT, Stoodley N, Moore AT, Wilson S, Maher ER. Mutation in Rab3 GTPase-activating protein (RAB3GAP) noncatalytic subunit in a kindred with Martsolf syndrome. Am J Hum Genet. 2006;78:702–7. [PMC free article: PMC1424696] [PubMed: 16532399]
- Arroyo-Carrera I, de Zaldívar Tristancho MS, Bermejo-Sánchez E, Martínez-Fernández ML, López-Lafuente A, MacDonald A, Zúñiga Á, Luis Gómez-Skarmeta J, Luisa Martínez-Frías M. Deletion 1q43-44 in a patient with clinical diagnosis of Warburg-Micro Syndrome. Am J Med Genet. 2015;167:1243–51. [PubMed: 25899426]
- Asahina M, Endoh Y, Matsubayashi T, Fukuda T, Ogata T. Novel RAB3GAP1 compound heterozygous mutations in Japanese siblings with Warburg micro syndrome. Brain Dev. 2016;38:337–40. [PubMed: 26421802]
- Bem D, Yoshimura S, Nunes-Bastos R, Bond FC, Kurian MA, Rahman F, Handley MT, Hadzhiev Y, Masood I, Straatman-Iwanowska AA, Cullinane AR, Mcneill A, Pasha SS, Kirby GA, Foster K, Ahmed Z, Morton JE, Williams D, Graham JM, Dobyns WB, Burglen L, Ainsworth JR, Gissen P, Muller F, Maher ER, Barr FA, Aligianis IA. Loss-of-function mutations in RAB18 cause Warburg micro syndrome. Am J Hum Genet. 2011;88:499–507. [PMC free article: PMC3071920] [PubMed: 21473985]
- Borck G, Wunram H, Steiert A, Volk AE, Korber F, Roters S, Herkenrath P, Wollnik B, Morris-Rosendahl DJ, Kubisch C. A homozygous RAB3GAP2 mutation causes Warburg micro syndrome. Hum Genet. 2011;129:45–50. [PubMed: 20967465]
- Clabecq A, Henry JP, Darchen F. Biochemical characterization of Rab3-GTPase-activating protein reveals a mechanism similar to that of Ras-GAP. J Biol Chem. 2000;275:31786–91. [PubMed: 10859313]
- Dejgaard SY, Murshid A, Erman A, Kizilay O, Verbich D, Lodge R, Dejgaard K, Ly-Hartig TB, Pepperkok R, Simpson JC, Presley JF. Rab18 and Rab43 have key roles in ER-Golgi trafficking. J Cell Sci. 2008;121:2768–81. [PubMed: 18664496]
- Dursun F, Guven A, Morris-Rosendahl D. Warburg micro syndrome. J Pediatr Endocrinol Metab. 2012;25:379–82. [PubMed: 22768674]
- Feldmann A, Bekbulat F, Huesmann H, Ulbrich S, Tatzelt J, Behl C, Kern A. The RAB GTPase RAB18 modulates macroautophagy and proteostasis. Biochem Biophys Res Commun. 2017;486:738–43. [PubMed: 28342870]
- Frasa MA, Koessmeier KT, Ahmadian MR, Braga VM. Illuminating the functional and structural repertoire of human TBC/RABGAPs. Nat Rev Mol Cell Biol. 2012;13:67–73. [PubMed: 22251903]
- Gavriljuk K, Gazdag EM, Itzen A, Kotting C, Goody RS, Gerwert K. Catalytic mechanism of a mammalian Rab.RabGAP complex in atomic detail. Proc Natl Acad Sci U S A. 2012;109:21348–53. [PMC free article: PMC3535612] [PubMed: 23236136]
- Gerondopoulos A, Bastos RN, Yoshimura S, Anderson R, Carpanini S, Aligianis I, Handley MT, Barr FA. Rab18 and a Rab18 GEF complex are required for normal ER structure. J Cell Biol. 2014;205:707–20. [PMC free article: PMC4050724] [PubMed: 24891604]
- Gillespie RL, O'Sullivan J, Ashworth J, Bhaskar S, Williams S, Biswas S, Kehdi E, Ramsden SC, Clayton-Smith J, Black GC, Lloyd IC. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology. 2014;121:2124-37.e1-2. [PubMed: 25148791]
- Graham JM Jr, Hennekam R, Dobyns WB, Roeder E, Busch D. MICRO syndrome: an entity distinct from COFS syndrome. Am J Med Genet A. 2004;128A:235–45. [PubMed: 15216543]
- Gupta NTS, Thakur S, Handley MT, Bokaria R, Saxena R, Kohli S. A genetic syndrome that mimics congenital TORCH infection. Genetic Clinics. 2016. Available online. Accessed 2-23-22.
- Haas AK, Yoshimura S, Stephens DJ, Preisinger C, Fuchs E, Barr FA. Analysis of GTPase-activating proteins: Rab1 and Rab43 are key Rabs required to maintain a functional Golgi complex in human cells. J Cell Sci. 2007;120:2997–3010. [PubMed: 17684057]
- Handley MT. RAB18. In: Choi S, ed. Encyclopedia of Signaling Molecules. 2 ed. New York, NY: Springer. 2017.
- Handley MT, Carpanini SM, Mali GR, Sidjanin DJ, Aligianis IA, Jackson IJ, Fitzpatrick DR. Warburg micro syndrome is caused by RAB18 deficiency or dysregulation. Open Biol. 2015;5:150047. [PMC free article: PMC4632505] [PubMed: 26063829]
- Handley MT, Morris-Rosendahl DJ, Brown S, Macdonald F, Hardy C, Bem D, Carpanini SM, Borck G, Martorel L, Izzi C, Faravelli F, Accorsi P, Pinelli L, Basel-Vanagaite L, Peretz G, Abdel-Salam GM, Zaki MS, Jansen A, Mowat D, Glass I, Stewart H, Mancini G, Lederer D, Roscioli T, Giuliano F, Plomp AS, Rolfs A, Graham JM, Seemanova E, Poo P, Garcia-Cazorla A, Edery P, Jackson IJ, Maher ER, Aligianis IA. Mutation spectrum in RAB3GAP1, RAB3GAP2, and RAB18 and genotype-phenotype correlations in Warburg micro syndrome and Martsolf syndrome. Hum Mutat. 2013;34:686–96. [PubMed: 23420520]
- Imagawa E, Fukai R, Behnam M, Goyal M, Nouri N, Nakashima M, Tsurusaki Y, Saitsu H, Salehi M, Kapoor S, Tanaka F, Miyake N, Matsumoto N. Two novel homozygous RAB3GAP1 mutations cause Warburg micro syndrome. Hum Genome Var. 2015;2:15034. [PMC free article: PMC4785564] [PubMed: 27081543]
- Irimia M, Weatheritt RJ, Ellis JD, Parikshak NN, Gonatopoulos-Pournatzis T, Babor M, Quesnel-Vallieres M, Tapial J, Raj B, O'Hanlon D, Barrios-Rodiles M, Sternberg MJ, Cordes SP, Roth FP, Wrana JL, Geschwind DH, Blencowe BJ. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell. 2014;159:1511–23. [PMC free article: PMC4390143] [PubMed: 25525873]
- Kabzińska D, Mierzewska H, Senderek J, Kochański A. Warburg micro syndrome type 1 associated with peripheral neuropathy and cardiomyopathy. Folia Neuropathol. 2016;54:273–81. [PubMed: 27764520]
- Leung KF, Baron R, Ali BR, Magee AI, Seabra MC. Rab GTPases containing a CAAX motif are processed post-geranylgeranylation by proteolysis and methylation. J Biol Chem. 2007;282:1487–97. [PubMed: 17114793]
- Liegel RP, Handley MT, Ronchetti A, Brown S, Langemeyer L, Linford A, Chang B, Morris-Rosendahl DJ, Carpanini S, Posmyk R, Harthill V, Sheridan E, Abdel-Salam GM, Terhal PA, Faravelli F, Accorsi P, Giordano L, Pinelli L, Hartmann B, Ebert AD, Barr FA, Aligianis IA, Sidjanin DJ. Loss-of-function mutations in TBC1D20 cause cataracts and male infertility in blind sterile mice and Warburg micro syndrome in humans. Am J Hum Genet. 2013;93:1001–14. [PMC free article: PMC3852926] [PubMed: 24239381]
- Mandarano R, Danieli A, Faletra F, Michieletto P, Montanaro D, Martinuzzi A, Handley MT, Bonanni P. "Myoclonic absences" and other novel findings in Warburg micro syndrome: clinical report of an expanding RAB18 phenotype. J Mol Genet Med. 2017. Available online. Accessed 2-23-22.
- Martin S, Driessen K, Nixon SJ, Zerial M, Parton RG. Regulated localization of Rab18 to lipid droplets: effects of lipolytic stimulation and inhibition of lipid droplet catabolism. J Biol Chem. 2005;280:42325–35. [PubMed: 16207721]
- Martsolf JT, Hunter AG, Haworth JC. Severe mental retardation, cataracts, short stature, and primary hypogonadism in two brothers. Am J Med Genet. 1978;1:291–9. [PubMed: 677168]
- Morris-Rosendahl DJ, Segel R, Born AP, Conrad C, Loeys B, Brooks SS, Muller L, Zeschnigk C, Botti C, Rabinowitz R, Uyanik G, Crocq MA, Kraus U, Degen I, Faes F. New RAB3GAP1 mutations in patients with Warburg micro syndrome from different ethnic backgrounds and a possible founder effect in the Danish. Eur J Hum Genet. 2010;18:1100–6. [PMC free article: PMC2987448] [PubMed: 20512159]
- Nassogne MC, Henrot B, Saint-Martin C, Kadhim H, Dobyns WB, Sebire G. Polymicrogyria and motor neuropathy in micro syndrome. Neuropediatrics. 2000;31:218–21. [PubMed: 11071150]
- Ozeki S, Cheng J, Tauchi-Sato K, Hatano N, Taniguchi H, Fujimoto T. Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum-derived membrane. J Cell Sci. 2005;118:2601–11. [PubMed: 15914536]
- Patel N, Anand D, Monies D, Maddirevula S, Khan AO, Algoufi T, Alowain M, Faqeih E, Alshammari M, Qudair A, Alsharif H, Aljubran F, Alsaif HS, Ibrahim N, Abdulwahab FM, Hashem M, Alsedairy H, Aldahmesh MA, Lachke SA, Alkuraya FS. Novel phenotypes and loci identified through clinical genomics approaches to pediatric cataract. Hum Genet. 2017;136:205–25. [PMC free article: PMC5783298] [PubMed: 27878435]
- Picker-Minh S, Busche A, Hartmann B, Spors B, Klopocki E, Hubner C, Horn D, Kaindl AM. Large homozygous RAB3GAP1 gene microdeletion causes Warburg micro syndrome 1. Orphanet J Rare Dis. 2014;9:113. [PMC free article: PMC4224754] [PubMed: 25332050]
- Pulido MR, Diaz-Ruiz A, Jimenez-Gomez Y, Garcia-Navarro S, Gracia-Navarro F, Tinahones F, Lopez-Miranda J, Fruhbeck G, Vazquez-Martinez R, Malagon MM. Rab18 dynamics in adipocytes in relation to lipogenesis, lipolysis and obesity. PLoS One. 2011;6:e22931. [PMC free article: PMC3145781] [PubMed: 21829560]
- Rump P, Jazayeri O, Van Dijk-Bos KK, Johansson LF, Van Essen AJ, Verheij JB, Veenstra-Knol HE, Redeker EJ, Mannens MM, Swertz MA, Alizadeh BZ, Van Ravenswaaij-Arts CM, Sinke RJ, Sikkema-Raddatz B. Whole-exome sequencing is a powerful approach for establishing the etiological diagnosis in patients with intellectual disability and microcephaly. BMC Med Genomics. 2016;9:7. [PMC free article: PMC4743197] [PubMed: 26846091]
- Sawyer SL, Schwartzentruber J, Beaulieu CL, Dyment D, Smith A, Warman Chardon J, Yoon G, Rouleau GA, Suchowersky O, Siu V, Murphy L, Hegele RA, Marshall CR, Bulman DE, Majewski J, Tarnopolsky M, Boycott KM, et al. Exome sequencing as a diagnostic tool for pediatric-onset ataxia. Hum Mutat. 2014;35:45–9. [PMC free article: PMC4255313] [PubMed: 24108619]
- Srivastava P, Saxena D, Joshi S, Phadke SR. Consanguinity as an adjunct diagnostic tool. Indian J Pediatr. 2016;83:258–60. [PubMed: 26138576]
- Tasdemir S, Sahin I, Morris-Rosendahl DJ, Marzioglu E, Cayir A, Yuce I, Tatar A. Recurrent Rab3gap1 mutations in the Turkish population. Genet Couns. 2015;26:415–23. [PubMed: 26852512]
- Trkova M, Hynek M, Dudakova L, Becvarova V, Hlozanek M, Raskova D, Vincent AL, Liskova P. Early detection of bilateral cataracts in utero may represent a manifestation of severe congenital disease. Am J Med Genet A. 2016;170:1843–8. [PubMed: 27256633]
- Vazquez-Martinez R, Cruz-Garcia D, Duran-Prado M, Peinado JR, Castano JP, Malagon MM. Rab18 inhibits secretory activity in neuroendocrine cells by interacting with secretory granules. Traffic. 2007;8:867–82. [PubMed: 17488286]
- Warburg M, Sjo O, Fledelius HC, Pedersen SA. Autosomal recessive microcephaly, microcornea, congenital cataract, mental retardation, optic atrophy, and hypogenitalism. Micro syndrome. Am J Dis Child. 1993;147:1309–12. [PubMed: 8249951]
- Yildirim MS, Zamani AG, Bozkurt B. Warburg micro syndrome in two children from a highly inbred Turkish family. Genet Couns. 2012;23:169–74. [PubMed: 22876574]
- Yüksel A, Yesil G, Aras C, Seven M. Warburg micro syndrome in a Turkish boy. Clin Dysmorphol. 2007;16:89–93. [PubMed: 17351351]
Chapter Notes
Acknowledgments
We thank the patients, their families, and clinicians for their collaboration and contribution to our knowledge about Warburg micro syndrome and Martsolf syndrome.
Revision History
- 4 January 2018 (bp) Review posted live
- 2 May 2017 (mh) Original submission
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: January 4, 2018.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Handley M, Sheridan E. RAB18 Deficiency. 2018 Jan 4. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.