Summary
Clinical characteristics.
X-linked congenital retinoschisis (XLRS) is characterized by symmetric bilateral macular involvement with onset in the first decade of life, in some cases as early as age three months. Fundus examination shows areas of schisis (splitting of the nerve fiber layer of the retina) in the macula, sometimes giving the impression of a spoke wheel pattern. Schisis of the peripheral retina, predominantly inferotemporally, occurs in approximately 50% of individuals. Affected males typically have 20/60 to 20/120 vision. Visual acuity often deteriorates during the first and second decades of life but then remains relatively stable until the fifth or sixth decade.
Diagnosis/testing.
The diagnosis of XLRS is established in a male proband with suggestive ophthalmologic findings and a hemizygous pathogenic variant in RS1 identified by molecular genetic testing.
Management.
Treatment of manifestations: Management of refractive errors and amblyopia is per standard care. Ambylopia prevention therapy is indicated following surgical intervention for vitreous hemorrhage or retinal detachment or in instances of severe retinoschisis or hypermetropia.
Low-vision aids such as large-print textbooks; preferential seating in the front of the classroom; and use of handouts with high contrast. Surgery may be required to address the infrequent complications of vitreous hemorrhage and full-thickness retinal detachment.
Surveillance: Annual evaluation of children younger than age ten years by a pediatric ophthalmologist or retina specialist; patient education and close follow up may allow for early identification of refractive errors and treatment of vision-threatening complications (retinal detachment).
Agents/circumstances to avoid: Head trauma and high-contact sports to reduce risk of retinal detachment and vitreous hemorrhage.
Genetic counseling.
XLRS is inherited in an X-linked manner. Heterozygous females have a 50% chance of transmitting the pathogenic variant in each pregnancy: males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant will be carriers and will nearly always have normal visual function. Affected males transmit the pathogenic variant to all of their daughters and none of their sons. Once the RS1 pathogenic variant in the family is known, carrier testing for at-risk female relatives, prenatal testing for pregnancies at increased risk, and preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
X-linked congenital retinoschisis (XLRS) should be suspected in males with following ophthalmologic findings and family history.
Ophthalmologic findings
Bilaterally reduced visual acuity, typically between 20/60 and 20/120
No presenting complaint of “night blindness” (i.e., vision difficulty in dim lighting)
Fundus examination revealing:
Spectral domain optical coherence tomography (SD-OCT), currently the major diagnostic technique for XLRS, reveals characteristic intraretinal foveal schisis in younger men, while cystic spaces become less evident by middle age as the flattening of cysts (with the appearance of partial macular atrophy) occurs with age [
Molday et al 2012].
Electroretinogram (ERG) in more than half of affected males shows characteristic changes of greatly diminished dark-adapted b-wave amplitude despite relative preservation of the a-wave amplitude, termed an "electronegative ERG" [
Nakamura et al 2001]. However, as some boys have a technically normal ERG b-wave [
Sieving et al 1999,
Eksandh et al 2005,
Renner et al 2008], the ERG may give false negative results when relatively normal. Note: Progressive reduction of ERG b-wave amplitudes can occur in older men with severe pathogenic variants (e.g., null variants, splice variants, cysteine conformational disruptions) [
Bowles et al 2011].
Intravenous fluorescein angiogram appears normal in younger boys, with no apparent leakage, whereas older individuals may have atrophic changes and involvement of the retinal pigment epithelium.
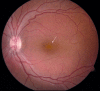
Fundus photo of a male with congenital retinoschisis. Arrow points to typical spoke-wheel pattern of foveal cysts.
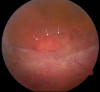
Fundus photo of the peripheral retina of a male with congenital retinoschisis. Area marked with arrows shows a partial-thickness retinal hole.
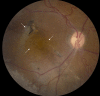
Fundus photo of a male with congenital retinoschisis showing atypical, more severe findings with arrows pointing to atrophic macular changes
Family history consistent with X-linked inheritance (e.g., no male-to-male transmission). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of XLRS is established in a male proband with suggestive ophthalmologic findings and a hemizygous pathogenic (or likely pathogenic) variant in RS1 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of a hemizygous RS1 variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive
genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of XLRS has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of RS1 is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
Note: Targeted analysis can be performed first for the following pathogenic variants in individuals of Finnish ancestry [Huopaniemi et al 1999]:
An inherited retinal dystrophy multigene panel that includes RS1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see Table 1).
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive
genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in X-Linked Congenital Retinoschisis
View in own window
| Gene 1 | Method | Proportion of Pathogenic Variants 2 Identified by Method |
|---|
|
RS1
| Sequence analysis 3 | 90%-95% 4 |
| Gene-targeted deletion/duplication analysis 5 | 5%-10% 4 |
- 1.
- 2.
- 3.
Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Variants may include missense, nonsense, and splice site variants and small intragenic deletions/insertions; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
- 4.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
Clinical Characteristics
Clinical Description
X-linked congenital retinoschisis (XLRS) is a symmetric bilateral macular disorder with onset in the first decade of life in males, and, in some instances, as early as age three months. Affected males generally present with reduction in vision by early elementary school. Affected males typically have vision of 20/60 to 20/120 on first presentation.
Visual acuity may deteriorate slightly during the first and second decades of life but then remains relatively stable until the fifth or sixth decade, when slowly progressive macular atrophy can occur [Eksandh et al 2000, Apushkin et al 2005]. Visual loss may later progress to legal blindness (acuity <20/200). Some men show macular pigmentary changes after age 50 years; some degree of atrophy of the retinal pigment epithelium is common.
Variations in disease severity and progression are observed even among members of the same family.
Appearance of foveal lesions varies from largely radial striations (3%), microcystic lesions (34%), honeycomb-like cysts (8%), or their combinations (31%) to non-cystic-appearing foveal changes including pigment mottling (8%), loss of the foveal reflex (8%), or an atrophic-appearing lesion (8%) [Apushkin et al 2005].
XLRS progresses to retinal detachment in an estimated 5% to 22% of affected individuals. Retinal detachment can occur in infants with severe retinoschisis. About 4% to 40% of individuals with XLRS develop vitreous hemorrhage.
Heterozygous (carrier) females. In most instances, heterozygous females cannot be identified by clinical examination as they nearly always have a normal fundus appearance and visual function. Very rarely, examination of the peripheral retina may show white flecks or areas of schisis.
Genotype-Phenotype Correlations
The phenotype comprises a spectrum ranging from a few schisis cavities at the mild end to major retinoschisis detachment (not surgically repairable) at the severe end. The following general genotype-phenotype correlations have been reported:
Molecular modeling indicates that predicted change and/or damage to retinoschisin determines progression rate and clinical severity as evaluated by ERG. Importantly, molecular alterations that result in nonsense, splice site, and cysteine disulfide bond variants appear to be associated with the most severe functional deterioration [Sergeev et al 2010, Bowles et al 2011, Sergeev et al 2013].
Nomenclature
Other terms correctly used in the past to refer to X-linked congenital retinoschisis:
X-linked juvenile retinoschisis
Juvenile macular degeneration/dystrophy
Degenerative retinoschisis
Vitreous veils of the retina
Other terms incorrectly used in the past to refer to X-linked congenital retinoschisis:
Cone dystrophy
Macular hole
Prevalence
According to the Retinoschisis Consortium [1998] (which comprises the largest collection of known cases of XLRS) the estimated prevalence ranges from 1:5,000 to 1:25,000.
Differential Diagnosis
While the presence of retinoschisis in an individual with a positive family history of X-linked congenital retinoschisis (XLRS) establishes the diagnosis in that person, making the diagnosis in a male with no known family history may be more difficult.
Hereditary Disorders
Table 2.
Genes of Interest in the Differential Diagnosis of X-Linked Congenital Retinoschisis
View in own window
| Gene | Disorder | MOI | Clinical Features of Differential Diagnosis Disorder | Distinguishing Features |
|---|
CACNA1F
NYX
|
X-linked congenital stationary night blindness
| XL | Electronegative ERG may mimic XLRS. | XLRS rarely presents w/complaint of "night blindness." |
|
NR2E3
| Goldmann-Favre vitreoretinal degeneration & enhanced S-cone syndrome (OMIM 268100) | AR | May mimic XLRS. Onset in infancy. Severely impaired vision incl marked visual field loss & severe night blindness. Coarse intraretinal cysts may be seen w/peripheral retinoschisis; no vitreous veils are observed. | ERG shows markedly ↓ a-waves & b-waves w/altered timing (vs simply the ↓ in the b-wave amplitude seen in XLRS). |
OFD1
RP2
RPGR
(>80 genes) 1 |
Nonsyndromic retinitis pigmentosa
| XL 1
(AD,
AR,
digenic) | RP: a group of inherited disorders involving congenital or progressive death of retinal photoreceptors (rods & cones) → visual loss. 2 Referring diagnosis in many persons w/XLRS (XLRP may cause confusion w/XLRS.) | Unlike XLRS, RP may be assoc w/intraretinal pigment dispersion or clumping, narrowing of retinal vessels, & optic nerve gliotic pallor. ERG in RP (esp XLRP) has markedly ↓ a-wave & b-wave (vs selective b-wave amplitude ↓ in XLRS) |
|
VCAN
| VCAN vitreoretinopathy
(Wagner syndrome & erosive vitreoretinopathy) (OMIM 143200) | AD | "Optically empty vitreous" on slit-lamp exam & avascular vitreous strands & veils, mild or occasionally moderate-to-severe myopia, presenile cataract, night blindness of variable degree assoc w/progressive chorioretinal atrophy, retinal traction & retinal detachment at advanced stages of disease, & ↓ visual acuity. 1st signs usually appear in early adolescence. | |
AD = autosomal dominant; AR = autosomal recessive; ERG = electroretinogram; MOI = mode of inheritance; XL = X-linked; XLRP = X-linked retinitis pigmentosa; XLRS = X-linked congenital retinoschisis
- 1.
- 2.
Noble et al [1978] reported a family with rod-cone dystrophy and associated foveal schisis. For this reason, foveal retinoschisis alone does not make the diagnosis of XLRS.
Acquired Disorders and Hereditary Disorders of Unknown Genetic Cause
Cystoid macular edema may mimic foveal retinoschisis. Macular edema may be caused by diabetes mellitus, inflammatory conditions of the eye (uveitis), or intraocular surgery.
Bilateral amblyopia can be a referring diagnosis when foveal schisis changes are subtle and difficult to observe. Suspicion of XLRS is raised if family history indicates other affected males in an X-linked inheritance pattern.
Degenerative retinoschisis is an idiopathic, degenerative disease of the peripheral retina. No evidence suggests genetic etiology [Lewis 2003]. Degenerative peripheral retinoschisis is primarily age related, with splitting in the outer retina through the outer nuclear layer and plexiform layer; in XLRS splitting may also involve the proximal retinal layers [Sieving 1998].
Retinal detachment, in which the full-thickness retina elevates and lifts off from the underlying ocular support, differs from retinoschisis, in which the retina splits through the nerve fiber layer. Retinal detachment in an otherwise normal eye can be surgically repaired, whereas retinal detachment associated with retinoschisis usually cannot.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with X-linked congenital retinoschisis (XLRS), the following evaluations (if not performed as part of the evaluation that led to the diagnosis) are recommended:
Complete ophthalmologic examination including:
Visual fields, by Goldmann or other perimetry
Fundoscopic examination
Optical coherence tomography
Electroretinogram; confirmatory for half or more of cases by an electronegative configuration or b-wave amplitude reduction disproportionate to a-wave loss
Consultation with a medical geneticist, certified genetic counselor, or certified advanced genetic nurse for the purpose of informing affected individuals and their families about the nature, mode of inheritance, and implications of XLRS in order to facilitate medical and personal decision making
Note:
Turriff et al [2020] interviewed and surveyed parents of sons following a confirmed diagnosis of XLRS and describe ways in which medical professionals can optimally support affected individuals and their families.
Treatment of Manifestations
Refractive errors and amblyopia. Management is per standard care.
Note that amblyopia prevention therapy is indicated following surgical intervention to treat vitreous hemorrhage or retinal detachment, or in cases of severe retinoschisis or hypermetropia [Kyung & Lee 2012].
Surgical intervention. Because retinoschisis affects primarily the inner retinal layers, retinoschisis alone (without retinal detachment) is at best difficult to treat surgically.
Care of a retinal surgeon may be required to address the infrequent complications of vitreous hemorrhage and full-thickness retinal detachment. Because the clinical presentation of a large area of peripheral retinoschisis may mask a true retinal detachment, consultation with an ophthalmologist or retinal surgeon is recommended when in doubt.
Low-vision services are designed to benefit those whose ability to function is compromised by vision impairment. Low-vision specialists, often optometrists, help optimize the use of remaining vision. Services provided vary based on age and needs.
Public school systems are mandated by federal law to provide appropriate education for children who have vision impairment.
Individualized education plan (IEP) services:
An IEP provides specially designed instruction and related services to children who qualify.
IEP services will be reviewed annually to determine whether any changes are needed.
Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
Vision consultants should be a part of the child's IEP team to support access to academic material.
A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
Other. Many individuals with XLRS are able to obtain a restricted driver's license. Some individuals have found specially designed telescopic lenses useful when driving; legal use of telescopic lenses may vary by locale.
Surveillance
Annual evaluation of children younger than age ten years by a pediatric ophthalmologist to diagnose refractive errors or by a retina specialist to examine the peripheral retina for schisis or detachment is recommended.
Older children and adults need less frequent monitoring as they would be more apt to report changes in vision.
Patient education and close follow up are the only clinical options that may allow for early identification and treatment of vision-threatening complications such as retinal detachment (Orphanet, accessed 11-2-20).
Agents/Circumstances to Avoid
Although retinal detachment and vitreous hemorrhage occur in a minority of affected individuals (5%-22% and 4%-40%, respectively), generally avoiding head trauma and high-contact sports is recommended.
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk male relatives of an affected individual in order to identify as early as possible those who would benefit from institution of treatment for retinal detachment and preventive measures (see Agents/Circumstances to Avoid).
If the RS1 pathogenic variant in the family is known, molecular genetic testing can be used to clarify the genetic status of at-risk male relatives.
If the pathogenic variant in the family is not known, examination by an ophthalmologist can clarify the affected status of at-risk relatives.
See Genetic Counseling for issues related to testing of at-risk male relatives for genetic counseling purposes.
Therapies Under Investigation
Successful ocular gene therapy has been demonstrated in mouse XLRS models [Zeng et al 2004, Min et al 2005].
Two human XLRS gene therapy trials were initiated in 2015 (National Eye Institute: ClinicalTrials.gov NCT02317887; AGTC, Inc: ClinicalTrials.gov NCT02416622):
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Note: See Turriff et al [2019] for discussion of how medical professionals can support informed decision making in individuals considering participation in a gene therapy trial.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
X-linked congenital retinoschisis (XLRS) is inherited in an X-linked manner.
Risk to Family Members
Parents of a male proband
The father of an affected male will not have the disorder nor will he be hemizygous for the RS1 pathogenic variant; therefore, he does not require further evaluation/testing.
In a family with more than one affected individual, the mother of an affected male is an obligate heterozygote (carrier). Note: If a woman has more than one affected son and no other affected relatives, and if the RS1 pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism.
Heterozygous females do not exhibit any signs in the macula, and only rarely are peripheral retinal changes associated with the carrier state.
If a male is the only affected family member (i.e., a simplex case), the mother may be a carrier or the affected male may have a
de novo
RS1 pathogenic variant, in which case the mother is not a carrier.
De novo pathogenic variants have been reported but are rare [
Gehrig et al 1999,
Kim et al 2009].
Sibs of a male proband. The risk to sibs depends on the genetic status of the mother:
If the mother of the proband has an RS1 pathogenic variant, the chance of transmitting it in each pregnancy is 50%:
Females who inherit the pathogenic variant will be carriers. Carriers nearly always have normal ocular examination, visual function, and electrophysiology (i.e., ERG).
If the proband represents a simplex case (i.e., a single occurrence in a family) and if the RS1 pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs of inheriting an RS1 pathogenic variant is greater than that of the general population because of the possibility of maternal germline mosaicism; the risk of maternal germline mosaicism in XLRS is not known.
Offspring of a male proband. Affected males transmit the RS1 pathogenic variant to:
Other family members. A male proband's maternal aunts are at risk of being carriers for the RS1 pathogenic variant and the aunts' offspring, depending on their sex, are at risk of being carriers for the pathogenic variant or of being affected.
Note: Molecular genetic testing may be able to identify the family member in whom a de novo pathogenic variant arose, thus providing information that could help determine genetic risk status of the extended family.
Carrier Detection
Molecular genetic testing of at-risk female relatives to determine their genetic status is most informative if the RS1 pathogenic variant has been identified in the proband.
Note: (1) Females who are carriers for this X-linked disorder nearly always have normal visual function and normal electrophysiology (i.e., ERG). (2) Identification of female heterozygotes requires either (a) prior identification of the RS1 pathogenic variant in the family or, (b) if an affected male is not available for testing, molecular genetic testing first by sequence analysis, and if no pathogenic variant is identified, by gene-targeted deletion/duplication analysis.
Prenatal Testing and Preimplantation Genetic Testing
Once the RS1 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
X-Linked Congenital Retinoschisis: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
RS1 encodes retinoschisin, a protein highly expressed within the inner segments of the photoreceptors that has a complex interaction within cells of the retina [Molday et al 2001]. Retinoschisin is a scaffold protein necessary for retinal architecture and organization, as well as maintenance of the photoreceptor-bipolar synapse. Further refinement in the understanding of protein function and interaction continues to be elucidated [Bush et al 2015, Strupaitė et al 2018, Plössl et al 2019, Ziccardi et al 2019].
Mechanism of disease causation. Loss of function
Table 3.
Notable RS1 Pathogenic Variants
View in own window
| Reference Sequences | DNA Nucleotide Change | Predicted Protein Change | Comment [Reference] |
|---|
NM_000330.3
NP_000321.1
| c.214G>A | p.Glu72Lys | Founder variants in the Finnish population [Huopaniemi et al 1999] |
| c.221G>T | p.Gly74Val |
| c.325G>C | p.Gly109Arg |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Chapter Notes
Author History
Stephanie Hoang, MSc (2014-present)
Ian M MacDonald, MD, CM (2003-present)
Paul A Sieving, MD, PhD (2003-present)
Meira Rina Meltzer, MA, MS; National Institutes of Health (2003-2014)
Nizar Smaoui, MD, FACMG; GeneDx (2003-2014)
Revision History
5 November 2020 (bp) Comprehensive update posted live
28 August 2014 (me) Comprehensive update posted live
12 May 2009 (me) Comprehensive update posted live
18 January 2006 (me) Comprehensive update posted live
24 October 2003 (me) Review posted live
1 July 2003 (ps) Original submission