Summary
Clinical characteristics.
Autosomal dominant Robinow syndrome (ADRS) is characterized by skeletal findings (short stature, mesomelic limb shortening predominantly of the upper limbs, and brachydactyly), genital abnormalities (in males: micropenis / webbed penis, hypoplastic scrotum, cryptorchidism; in females: hypoplastic clitoris and labia majora), dysmorphic facial features (widely spaced and prominent eyes, frontal bossing, anteverted nares, midface retrusion), dental abnormalities (including malocclusion, crowding, hypodontia, late eruption of permanent teeth), bilobed tongue, and occasional prenatal macrocephaly that persists postnatally. Less common findings include renal anomalies, radial head dislocation, vertebral abnormalities such as hemivertebrae and scoliosis, nail dysplasia, cardiac defects, cleft lip/palate, and (rarely) cognitive delay. When present, cardiac defects are a major cause of morbidity and mortality.
A variant of Robinow syndrome, associated with osteosclerosis and caused by a heterozygous pathogenic variant in DVL1, is characterized by normal stature, persistent macrocephaly, increased bone mineral density with skull osteosclerosis, and hearing loss, in addition to the typical features described above.
Management.
Treatment of manifestations: Corrective surgeries as needed for cryptorchidism, abnormal penile insertion / penoscrotal position, and cleft lip/palate. Hormone therapy may be helpful for males with micropenis. Orthodontic treatment is typically required.
Surveillance: Measurement of head circumference regularly in infancy and throughout childhood. Developmental assessment every three months in infancy and every six months to one year thereafter, or more frequently as needed if cognitive delays are identified. Dental evaluation every six to 12 months or as recommended. Periodic hearing assessments in childhood. Regular cardiac and renal assessment as needed by respective specialists if abnormalities are identified.
Evaluation of relatives at risk: Evaluation of the sibs of a proband in order to identify as early as possible those who would benefit from institution of treatment and surveillance.
Pregnancy management: Pregnancy in affected women appears to be generally uncomplicated. For an affected fetus, cesarean section may be required for abnormal presentation and/or cephalopelvic disproportion.
Genetic counseling.
ADRS is inherited in an autosomal dominant manner. A proband may have the disorder as a result of either an inherited or de novo pathogenic variant. Each child of an individual with ADRS has a 50% chance of inheriting the pathogenic variant; however, the severity of the clinical manifestations cannot be predicted from the results of molecular genetic testing. Prenatal testing for a pregnancy at increased risk is possible if the DVL1, DVL3, or WNT5A pathogenic variant has been identified in an affected family member.
Diagnosis
Suggestive Findings
Autosomal dominant Robinow syndrome (ADRS) should be suspected in individuals with the following clinical and family history findings [Mazzeu et al 2007, Person et al 2010, Beiraghi et al 2011, Roifman et al 2015].
Clinical Findings
Skeletal
Genital
In males: micropenis / webbed penis, hypoplastic scrotum, and cryptorchidism
In females: hypoplastic clitoris and labia majora
Craniofacial
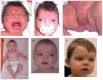
Dysmorphic facial features resembling a fetal face: widely spaced and prominent eyes, high anterior hairline, frontal bossing, depressed nasal bridge, short nose with anteverted nares, wide nasal bridge with a broad nasal tip, long philtrum, midface retrusion, and low-set ears (See and .)
Dental malocclusion, dental crowding and hypodontia, late eruption of permanent teeth, wide retromolar ridge, alveolar ridge deformation, and bilobed tongue
A mother and son, both affected with WNT5A-associated autosomal dominant Robinow syndrome A. Affected mother in infancy
A boy with WNT5A-associated autosomal dominant Robinow syndrome at different ages. Note the widely spaced and prominent eyes, high anterior hairline, frontal bossing, depressed nasal bridge, short nose with anteverted nares, wide nasal bridge with a broad (more...)
Family History
Family history is consistent with autosomal dominant inheritance. Note: Absence of a known family history of autosomal dominant Robinow syndrome does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of autosomal dominant Robinow syndrome is established in a proband with typical suggestive findings and/or by the identification of a heterozygous pathogenic (or likely pathogenic) variant in DVL1, DVL3, or WNT5A through molecular genetic testing (see Table 1).
Note: (1) If a heterozygous pathogenic variant is not identified in DVL1, DVL3, or WNT5A, it is appropriate to exclude the presence of biallelic ROR2 or NXN pathogenic variants (which cause autosomal recessive Robinow syndrome) and a heterozygous pathogenic variant in FZD2 (which causes autosomal dominant omodysplasia type 2). (2) Per ACMG variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants.
Molecular genetic testing approaches can include a combination of gene-targeted testing (concurrent or serial single-gene testing, multigene panel) and comprehensive
genomic testing (exome sequencing, genome sequencing):
Table 1.
Molecular Genetic Testing Used in Autosomal Dominant Robinow Syndrome
View in own window
| Gene 1, 2 | Proportion of Autosomal Dominant Robinow Syndrome Attributed to Pathogenic Variants in Gene | Proportion of Pathogenic Variants 3 Detectable by Sequence Analysis 4, 5 |
|---|
|
DVL1
| 18 probands 6 (unknown number of probands tested) | >99% 7 |
|
DVL3
| 7 probands 8 (unknown number of probands tested) | >99% 7 |
|
WNT5A
| 8 probands 9 (unknown number of probands tested) | >99% |
| Unknown 10, 11 | | NA |
- 1.
Genes are listed in alphabetic order.
- 2.
- 3.
- 4.
- 5.
Since ADRS occurs through a gain-of-function mechanism and large intragenic deletion or duplication has not been reported, testing for intragenic deletions or duplications is unlikely to identify a disease-causing variant.
- 6.
- 7.
All pathogenic variants identified to date are frameshift variants.
- 8.
- 9.
- 10.
Pathogenic variants in RAC3 and GPC4 have also been reported in single individuals who had been given a clinical diagnosis of Robinow syndrome [White et al 2018].
- 11.
Omodysplasia type 2, caused by pathogenic variants in FZD2, shares many clinical features with ADRS; it is unclear if these two conditions are part of a phenotypic spectrum (see Differential Diagnosis).
Clinical Characteristics
Clinical Description
Autosomal dominant Robinow syndrome (ADRS) is a skeletal dysplasia in which affected individuals typically have short stature, mesomelic limb shortening (predominantly of the upper limbs), and brachydactyly. A variety of other (variably present) anomalies may also suggest the diagnosis.
Facial. Craniofacial features of ADRS are summarized in Suggestive Findings. These features are most recognizable at birth or in early childhood. The distinctive facial features become less apparent with age.
In addition to macrocephaly, prominent facial features in adulthood include widely spaced eyes, wide nasal bridge, and broad nasal tip.
Dental malocclusion becomes apparent in early childhood and persists into adulthood, affecting the permanent dentition as well. One case of persistent primary dentition requiring extraction at age 18 years has been reported [
Roifman et al 2015].
Skeletal. Short stature is almost always present at birth and sometimes identified prenatally (on detailed fetal ultrasound) as early as age 20 weeks [Mazzeu et al 2007, Castro et al 2014, Roifman et al 2015].
Urogenital. Hypoplastic genitalia are apparent at birth for males and females.
Puberty and fertility. To the best of the authors' knowledge, both puberty and fertility are normal and affected females can carry pregnancies to term; delivery may need to be by cesarean section because of cephalopelvic disproportion.
Cardiac abnormalities occur in a minority (<25%) of individuals with ADRS.
Hearing loss (bilateral, mixed) has been reported in some individuals with DVL1-associated ADRS [Bunn et al 2015, White et al 2015].
Umbilical hernia has been reported in some individuals with DVL1-associated ADRS [White et al 2015].
Intelligence is usually normal; cognitive delay occurs in a minority of individuals with ADRS.
Other features less frequently seen (<25% of cases) [Mazzeu et al 2007, Person et al 2010, Roifman et al 2015]:
Renal anomalies (usually hydronephrosis)
Radial head dislocation
Vertebral abnormalities and scoliosis
Persistent primary teeth requiring extraction
Nail dysplasia
Cleft lip/palate
Phenotype Correlations by Gene
DVL1. A subset of individuals with DVL1-associated ADRS exhibit a final stature in the low-normal range, increased bone mineral density with osteosclerosis of the skull, and macrocephaly (ranging from +2.5SD to >+6SD) [Bunn et al 2015, White et al 2015].
DVL3. Three out of four individuals with DVL3-associated ADRS had cardiac abnormalities; osteosclerosis has not been reported.
WNT5A. Five families with WNT5A-associated ADRS harbor domain-specific pathogenic variants in the WNT5A protein and may represent a clinical phenotype with classic ADRS features (with characteristic face, short stature, mesomelic limb shortening, and genital hypoplasia) [Roifman et al 2015].
Prevalence
ADRS is very rare. The exact prevalence of the disorder is unknown. Fewer than 80 families with ADRS have been described in the literature.
Differential Diagnosis
ROR2-related Robinow syndrome is an autosomal recessive skeletal dysplasia caused by biallelic pathogenic variants in ROR2. Features similar to those of ADRS include the distinctive fetal face features, short stature, mesomelic limb shortening, and genital hypoplasia. ROR2-related Robinow syndrome appears to be more severe than ADRS, with renal anomalies, congenital heart defects, vertebral defects, rib fusions, scoliosis, and cognitive delay occurring more frequently than in ADRS. A distinguishing feature of ROR2-related Robinow syndrome is clefting of the distal phalanges, mainly of the thumbs.
NXN-related Robinow syndrome (OMIM 618529), also inherited in an autosomal recessive manner, was described in three individuals with biallelic NXN pathogenic variants from two unrelated families. All three had classic clinical findings of Robinow syndrome including typical craniofacial features, mesomelic shortening, and brachydactyly [White et al 2018]. One individual, born to consanguineous parents, was homozygous for a nonsense NXN variant; the two affected sibs in the other family had compound heterozygous NXN pathogenic variants.
Note: The NXN protein is a relevant partner in the WNT5A signaling pathway that is intimately involved in Robinow syndrome causation. ROR2 binds to WNT5A and interacts with FZD2. The effect of this interaction is routed to disheveled proteins (DVL1, DVL3) that are further stabilized by NXN. This complex activates JNK signaling responsible for cytoskeletal reorganization and cell polarity.
Aarskog syndrome (OMIM 305400) is an X-linked disorder caused by mutation of FGD1. Facial features (high anterior hairline, frontal bossing, widely spaced eyes, and anteverted nares) are similar to those of ADRS. Pulmonary valve stenosis has been reported. Genital hypoplasia in males with Aarskog syndrome is characterized by a shawl scrotum in contrast to the wider spectrum of genital hypoplasia found in ADRS. Other distinguishing features include widow's peak and ligamentous laxity. The vertebral abnormalities and delayed teeth eruption of ADRS are not observed in Aarskog syndrome. Typical limb abnormalities in Aarskog syndrome include brachydactyly, syndactyly, and fifth-finger clinodactyly.
Autosomal dominant Opitz G/BBB syndrome
(ADOS) and X-linked Opitz G/BBB syndrome
(XLOS) are associated with deletion in 22q11.2 and mutation of MID1, respectively. Similarities with ADRS include facial features (high anterior hairline, frontal bossing, widely spaced eyes, wide nasal bridge, anteverted nares) and genitourinary abnormalities (hypospadias, cryptorchidism, hypoplastic/bifid scrotum). XLOS is also characterized by laryngotracheoesophageal defects (not found in ADRS) and brain abnormalities, developmental delay, and cleft lip and/or palate (much more common in ADOS and XLOS [50% of affected individuals] than ADRS). ADOS and XLOS are not usually associated with short stature or mesomelic limb shortening.
Achondroplasia is an autosomal dominant disorder caused by mutation of FGFR3. Facial features characteristic of achondroplasia are similar to those of ADRS (macrocephaly, high anterior hairline and frontal bossing, depressed nasal bridge, pointed nose, and midface retrusion). Individuals with achondroplasia have a larger head circumference than those with ADRS, continued macrocephaly throughout life, and an increased incidence of hydrocephalus. Distinctive skeletal features in achondroplasia include trident appearance of the fingers, lumbar gibbus and hypotonia in infancy, hyperlordosis, bowing of the legs later in childhood, and more severe shortening of all long bones. Widely spaced eyes are not a feature of achondroplasia.
Omodysplasia type 2 (OMIM 164745) is a rare autosomal dominant disorder characterized by skeletal findings, hypoplastic male genitalia (including micropenis, hypospadias, and cryptorchidism), and dysmorphic facial features. Distinct features include normal stature, rhizomelic upper-limb shortening, shortened first metacarpals, and shortened humeri with hypoplastic condyles. The legs are normal. Facial features include frontal bossing, depressed nasal bridge with bifid nasal tip, and a long philtrum. Individuals with omodysplasia type 2 do not have widely spaced eyes. A heterozygous pathogenic variant in FZD2 was found in four families with omodysplasia type 2 [White et al 2018]. Given the phenotypic overlap between omodysplasia type 2 and Robinow syndrome, some have postulated that this may actually fall into the clinical spectrum of Robinow syndrome with predominant short humeri and radial head dislocation.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with autosomal dominant Robinow syndrome (ADRS), the evaluations summarized in Table 2 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 2.
Recommended Evaluations Following Initial Diagnosis in Individuals with Autosomal Dominant Robinow Syndrome
View in own window
| System/Concern | Evaluation | Comment |
|---|
|
Craniofacial
| Clinical assessment for presence of orofacial clefting | Consider referral to craniofacial team |
| Orthodontics consultation | For misaligned teeth or persistent primary dentition |
|
Ears
| Hearing assessment | |
|
Cardiovascular
| Echocardiogram | To evaluate for congenital heart defect |
|
Genitourinary
| Renal ultrasound | To assess for renal anomalies & hydronephrosis |
| Females: pelvic ultrasound | To evaluate for müllerian anomalies |
| Males: assessment for abnormal penile insertion / penoscrotal position & cryptorchidism | Urology consultation, as appropriate |
| If micropenis present, consider consultation w/endocrinologist for possible hormone therapy |
|
Musculoskeletal
| Radiographs of limbs, chest, vertebrae, & skull | To establish the extent of skeletal involvement |
|
Neurologic
| Assessment for developmental delay | Referral for formal neuropsychiatric/cognitive testing if present |
|
Other
| Consultation w/clinical geneticist &/or genetic counselor | |
Treatment of Manifestations
Table 3.
Treatment of Manifestations in Individuals with Autosomal Dominant Robinow Syndrome
View in own window
| Manifestation/Concern | Treatment | Considerations/Other |
|---|
| Cleft lip/palate | Surgical correction | Mgmt by multidisciplinary craniofacial team recommended |
| Misaligned teeth or persistent primary dentition | Standard orthodontic treatment | |
| Hearing loss | Standard treatment | See Hereditary Hearing Loss and Deafness Overview. |
| Congenital heart defects | Standard treatment per cardiologist &/or cardiothoracic surgery | |
| Cryptorchidism | Orchidopexy | |
| Abnormal penile insertion / penoscrotal position | Referral to urologist | Discussion of potential surgical correction |
| Micropenis | Consideration of hormonal therapy 1 | Referral to endocrinologist |
- 1.
Injection of human chorionic gonadotropin and testosterone improved penile length and testicular volume in three boys with severe micropenis [Soliman et al 1998].
Surveillance
Table 4.
Recommended Surveillance for Individuals with Autosomal Dominant Robinow Syndrome
View in own window
| System/Concern | Evaluation | Frequency |
|---|
|
Craniofacial
| Dental eval | Every 6 mos to 1 yr or per dental professional |
|
Ears
| Hearing assessment | In childhood |
|
Cardiovascular
| Standard monitoring in those w/cardiac involvement | Per cardiologist |
|
Genitourinary
| Standard monitoring in those w/renal involvement | Per urologist |
|
Neurologic
| Assessment of developmental progress | At each visit in childhood/adolescence |
Evaluation of Relatives at Risk
It is appropriate to evaluate the sibs of a proband in order to identify as early as possible those who would benefit from institution of treatment and surveillance. If the DVL1, DVL3, or WNT5A pathogenic variant in the family is known, molecular genetic testing can be used to clarify the genetic status of at-risk sibs.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Pregnancy in affected women appears to be generally uncomplicated. For an affected fetus, cesarean section may be required for abnormal presentation and/or cephalopelvic disproportion. Breech presentation requiring cesarean section has been reported in one case of ADRS [Roifman et al 2015].
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Robinow syndrome caused by pathogenic variants in DVL1, DVL3, or WNT5A is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
Sibs of a proband. The risk to the sibs of a proband depends on the genetic status of the proband's parents:
If a parent of the
proband is affected, the risk to the sibs is 50%. However, the severity of the clinical manifestations cannot be predicted from the results of
molecular genetic testing.
When the parents are clinically unaffected, the risk to the sibs of a
proband appears to be low.
If the
DVL1,
DVL3, or
WNT5A pathogenic variant found in the
proband cannot be detected in the leukocyte DNA of either parent, the risk to sibs is presumed to be slightly greater than that of the general population (though still <1%) because of the theoretic possibility of parental
germline mosaicism.
Offspring of a proband. Each child of an individual with ADRS has a 50% chance of inheriting the pathogenic variant; however, the severity of the clinical manifestations cannot be predicted from the results of molecular genetic testing.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected, the parent's family members may be at risk.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the DVL1, DVL3, or WNT5A pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible. However, the severity of clinical manifestations cannot be predicted from the results of molecular genetic testing.
Fetal ultrasound evaluation. Short long bones (measuring -2SD), macrocephaly, and typical facial features (widely spaced eyes, broad nasal bridge, and flattened frontal bone) can be detected by fetal ultrasound evaluation at approximately 20 weeks' gestation [Castro et al 2014, Roifman et al 2015].
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Autosomal Dominant Robinow Syndrome: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Table B.
View in own window
|
164975 | WINGLESS-TYPE MMTV INTEGRATION SITE FAMILY, MEMBER 5A; WNT5A |
|
180700 | ROBINOW SYNDROME, AUTOSOMAL DOMINANT 1; DRS1 |
|
601365 | DISHEVELLED 1; DVL1 |
|
601368 | DISHEVELLED 3; DVL3 |
|
616331 | ROBINOW SYNDROME, AUTOSOMAL DOMINANT 2; DRS2 |
|
616894 | ROBINOW SYNDROME, AUTOSOMAL DOMINANT 3; DRS3 |
Molecular Pathogenesis
The Wnt family of proteins regulates critical morphogenic events, including embryonic patterning, and cell differentiation, growth, and migration. Wnt signaling pathways fall into two categories: canonic, which involve β-catenin, and non-canonic, which are independent of β-catenin.
WNT5A, which functions in the both the canonic and non-canonic pathways, is a Wnt family member critical for developmental processes requiring cell migration (reviewed in Nishita et al [2010]). WNT5A is a known coreceptor of the orphan tyrosine kinase receptor, ROR2 [Oishi et al 2003, Mikels & Nusse 2006, Schambony & Wedlich 2007], which would explain the overlap in clinical phenotype of WNT5A- and ROR2-associated Robinow syndrome.
DVL1 and DVL3 encode segment polarity protein disheveled homolog DVL-1 (DVL1) and DVL-3, respectively, which are essential downstream mediators of Wnt signaling – specifically, the Wnt5a-ROR2 non-canonic pathway [White et al 2015, White et al 2016].
DVL1
Gene structure.
DVL1 comprises 15 exons.
Pathogenic variants. Thus far, all variants associated with ADRS are frameshift variants in exons 14 and 15 (see examples in Table 5) [Bunn et al 2015, White et al 2015, White et al 2016]. The proposed gain-of-function mechanism (see Abnormal gene product) suggests that these variants, located in the ultimate and penultimate exons, produce truncated transcripts that escape nonsense-mediated decay.
Table 5.
DVL1 Selected Pathogenic Variants
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.1505_1517del13 | p.His502ProfsTer143 |
NM_004421.2
NP_004412.2
|
| c.1508delC | p.Pro503ArgfsTer146 |
| c.1519delT | p.Trp507GlyfsTer142 |
| c.1529delG | p.Gly510ValfsTer139 |
| c.1562delC | p.Pro521HisfsTer128 |
| c.1570_1571delinsC | p.Phe524ProfsTer125 |
| c.1576_1583delinsG | p.Pro526AlafsTer121 |
| c.1615delA | p.Ser539AlafsTer110 |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product. DVL1 is a member of the disheveled family of intracellular scaffolding proteins, located downstream of the Wnt receptor. DVL1 plays a role in transducing both canonic and non-canonic Wnt signaling (OMIM 601365).
Abnormal gene product. The abnormal gene product resulting from a frameshift in exons 14 and 15 is thought to impair Wnt signaling via a gain-of-function or dominant-negative mechanism, which may explain the unique skeletal phenotype of normal stature, macrocephaly, and increased bone density [White et al 2015].
DVL3
Gene structure.
DVL3 comprises 15 exons.
Pathogenic variants. Similar to DVL1, the majority of variants associated with ADRS are frameshift variants in exons 14 and 15 (see examples in Table 6) [White et al 2015, White et al 2016]. Splice site variants at the intron 14 acceptor site have also been reported [White et al 2016]. The proposed gain-of-function mechanism (see Abnormal gene product) suggests that these variants, located in the ultimate and penultimate exons, produce truncated transcripts that escape nonsense-mediated decay.
Table 6.
DVL3 Selected Pathogenic Variants
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.1585delG | p.Ala592ProfsTer139 |
NM_004423.3
NP_004414.3
|
| c.1617delG | p.Gln539HisfsTer129 |
| c.1715-2A>G | p.? |
| c.1715-1G>A | p.? |
| c.1716delC | p.Ser573ValfsTer95 |
| c.1749delC | p.Ser583ArgfsTer85 |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product. DVL3 is a member of the disheveled family of intracellular scaffolding proteins, located downstream of the Wnt receptor. DVL3 plays a role in transducing both canonic and non-canonic Wnt signaling.
Abnormal gene product. The abnormal gene product resulting from a frameshift in exons 14 and 15 is thought to impair Wnt signaling via a gain-of-function or dominant-negative mechanism, which may explain the unique skeletal phenotype of normal stature, macrocephaly, and increased bone density [White et al 2015].
WNT5A
Gene structure.
WNT5A comprises five exons. Alternate splicing results in multiple transcript variants. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. Pathogenic missense variants (Table 7) as well as an in-frame duplication and an in-frame deletion/duplication have been identified in five families with autosomal dominant Robinow syndrome [Person et al 2010, Roifman et al 2015, White et al 2018].
Table 7.
WNT5A Selected Pathogenic Variants
View in own window
DNA Nucleotide Change
(Alias 1) | Predicted Protein Change
(Alias 1) | Reference Sequences |
|---|
| c.206G>A | p.Cys69Tyr |
NM_003392.4
NP_003383.2
|
| c.248G>C | p.Cys83Ser |
| c.257A>G 2 | p.Tyr86Cys |
c.544_545delinsTC
(544–545CT>TC) | p.Cys182Ser
(Cys182Arg) | |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Variant designation that does not conform to current naming conventions
- 2.
Two families had this variant.
Normal gene product. WNT5A is critical for developmental processes requiring cell migration [reviewed in Nishita et al 2010]. The three-dimensional structure of the WNT5A protein is still unknown.
The WNT5A signaling gradient in the limb bud has been shown to control limb elongation [Gao et al 2011]. The murine ortholog of WNT5A was also shown to control several steps in gonadal development of mice models [Tevosian 2012].
Abnormal gene product. Based on a WNT5A homology model using the experimentally solved structure of WNT8, Roifman et al [2015] showed that pathogenic variants found in all individuals with WNT5A-associated ADRS appear to be located on one side of the protein and may affect interactions with other proteins in the Wnt pathway by disrupting normal complex formation and/or intra-protein bonds. Although the mechanism of WNT5A-related disease is not known, the reports of disease-associated missense or in-frame variants clustering in specific locations of the protein could indicate altered protein function as a disease mechanism.