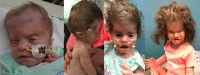
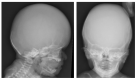
Clinical Description
SLC25A24 Fontaine progeroid syndrome is a multisystem connective tissue disorder characterized by poor growth, abnormal skeletal features, and distinctive craniofacial features with sagging, thin skin and decreased subcutaneous fat suggesting an aged appearance that is most pronounced in infancy and improves with time. Characteristic radiographic features include turribrachycephaly with widely open anterior fontanelle, craniosynostosis, and anomalies of the terminal phalanges. Cardiovascular, genitourinary, ocular, and gastrointestinal abnormalities may also occur.
Prior to identification of the molecular basis of this disorder, clinical findings were published by Gorlin et al [1960], Fontaine et al [1977], and Petty et al [1990]. Subsequent published case reports noted significant clinical overlap particularly with "Petty syndrome" and "Fontaine-Farriaux syndrome" [Castori et al 2009, Braddock et al 2010, Writzl et al 2017]. To date, 13 individuals have been described with a molecularly confirmed diagnosis of SLC25A24 Fontaine progeroid syndrome [Ehmke et al 2017; Writzl et al 2017; Rodríguez-García et al 2018; Ryu et al 2019; Legué et al 2020; Author, unpublished data]. The clinical findings discussed in this section are based on these reports (see Table 2).
Note: (1) Of the four individuals with an SLC25A24 pathogenic variant included in the report by Writzl et al [2017], three had previously been reported: Patient 2 in Faivre et al [1999], Patient 3 in Rodríguez et al [1999], and Patient 4 in Castori et al [2009]. (2) Of the five females with an SLC25A24 pathogenic variant and clinical findings suggestive of Gorlin-Chaudhry-Moss syndrome included in the report by Ehmke et al [2017], one (Patient 4) had previously been reported by Adolphs et al [2011].
Table 2.
Select Features of SLC25A24 Fontaine Progeroid Syndrome
View in own window
| Feature | Proportion
of Persons
w/Feature 1 | Comment |
|---|
| Pre- & postnatal growth failure | 12/13 | Poor weight gain is universal; short stature is observed in most persons. |
| Abnormal skull | 13/13 |
|
| Characteristic craniofacial features | 13/13 | See . |
| Ocular anomalies | 10/10 | Incl microphthalmia, hyperopia, eyelid anomalies, & blue or gray sclera |
| Sagging, thin, translucent skin | 12/12 | |
| Hypertrichosis | 10/10 | Involving face, back, & extensor surfaces of arms & legs |
| Skeletal anomalies | 13/13 |
|
| Cardiovascular anomalies | 9/11 | Structural abnormalities (5/11) Pulmonary hypertension (4/10) Aortic dilatation (6/11) Type A aortic dissection (1/11)
|
| External genital anomalies | 11/12 |
|
| Developmental delay w/normal cognition | 7/8 | Delayed acquisition of milestones, particularly motor skills, is common. In school-aged children, normal academic progress was reported in 7/8.
|
Growth
Prenatal growth. All but one affected individual presented with intrauterine growth restriction [Ehmke et al 2017]. Oligohydramnios is also commonly noted in the third trimester.
Z scores for birth weight ranged from -1 to -4.7, for birth length from +0.1 to -4, and for head circumference from -1.4 to -5.
Postnatal growth. Poor weight gain was observed in all individuals who lived more than 24 hours. Z scores for weight at time of last evaluation ranged from -2.7 to -6.
Feeding problems in infancy with poor growth are common. Three individuals have required gastrostomy feeds for poor feeding and/or reflux with aspiration [Ehmke et al 2017; Author, unpublished data].
Short stature was present in nine of 11 individuals who lived more than 24 hours, showing poor linear growth with Z scores ranging from -1.2 to -6.
Microcephaly, proportionate to height, is frequently present.
Craniofacial Features
Turribrachycephaly or brachycephaly is apparent at birth in most individuals and may be noted prenatally. In all but two individuals for whom there are detailed infant records, large anterior fontanelle is present. In the 11 individuals who lived longer than the neonatal period, craniosynostosis (most often involving the coronal sutures) was confirmed in nine and suspected in another.
Skin and Hair
Excessively wrinkled, sagging, and thin skin is typically noted at birth. Dermal translucence with prominent vasculature and decreased subcutaneous fat is common. In combination these skin findings (reflecting an apparent lipodystrophy) contribute to a progeroid appearance in infancy that improves with time.
The hair pattern is distinctive. Hypertrichosis, most frequently over the back, neck, and face, is present at birth and persists at least through childhood. In neonates, sparse frontotemporal scalp hair with low anterior and posterior hairlines are frequently noted. With age, scalp hair becomes coarse and abnormal hair whorls and growth patterns are common (see ).
Skeletal
Digital abnormalities, present in all reported individuals, include a mix of short distal phalanges (11/13), cutaneous syndactyly (7/13), and nail aplasia or hypoplasia (12/13) [Ehmke et al 2017; Writzl et al 2017; Rodríguez-García et al 2018; Ryu et al 2019; Legué et al 2020; Author, unpublished data]. Radiographs show shortened distal phalanges.
Mixed nail hypoplasia and aplasia may be present on some digits; normal nails may be present on other digits. Postaxial digits of hands and feet tend to be more severely affected than preaxial digits.
Cutaneous syndactyly of hands or feet was present in seven individuals; in the six for whom it was specified, syndactyly involved toes 2-3 and 4-5 (in 2 individuals) [Ehmke et al 2017; Author, unpublished data], toes 2-3 only (1 individual), toes 4-5 only (1 individual) [Ehmke et al 2017, Legué et al 2020], fingers 3-4 only (1 individual), and fingers 4-5 only (1 individual) [Writzl et al 2017, Ryu et al 2019].
Other skeletal findings have been variably reported and include the following:
Nonspecific long bone diaphyseal and ossification abnormalities in one infant [
Rodríguez et al 1999]
Cardiovascular
Congenital heart defects (patent ductus arteriosus, atrial septal defect secundum, bicuspid aortic valve, and dysplastic valves) occurred in four individuals [Ehmke et al 2017, Writzl et al 2017].
Pulmonary hypertension occurred in four individuals during infancy [Ehmke et al 2017; Writzl et al 2017; Author, unpublished data].
Two had normal neonatal echocardiograms and developed pulmonary hypertension before age six months [Writzl et al 2017; Author, unpublished data].
Gastroesophageal reflux and aspiration requiring fundoplication and gastrostomy tube occurred in at least two individuals who also developed pulmonary hypertension [Ehmke et al 2017; Author, unpublished data]. In at least one individual, right heart pressure improved following gastric fundoplication [Author, unpublished data].
Aortic dilatation was seen in six of the 11 individuals who lived more than 24 hours. Aortic dilatation, which had not been present on neonatal echocardiograms, developed in at least three individuals who did not have other cardiac anomalies [Ehmke et al 2017; Rodríguez-García et al 2018; Ryu et al 2019; Legué et al 2020; Author, unpublished data]. In one individual dilatation progressed during treatment with atenolol [Rodríguez-García et al 2018]. Another individual presented with an ascending aortic dissection at age 45 years [Legué et al 2020].
Development
Delayed acquisition of milestones in infancy is common. Normal cognition has been described in individuals living beyond early childhood.
Specific details regarding acquisition of developmental milestones are limited to four individuals in whom independent sitting was achieved at 12-18 months, walking at 19-36 months, and speech at 16-24 months [Ehmke et al 2017; Rodríguez-García et al 2018; Ryu et al 2019; Author, unpublished data].
Other
Central nervous system
Two individuals had hydrocephalus requiring shunt placement [
Ehmke et al 2017]; these were the only individuals with relative macrocephaly compared to height.
Two individuals had brain malformations: one with thin corpus callosum, ventriculomegaly, dysplastic cerebella vermis, pineal gland cyst, and large retrocerebellar area [
Ehmke et al 2017]; and one with gyral simplification consistent with pachygyria, cerebellar hypoplasia, and subependymal heterotopia noted on autopsy [
Writzl et al 2017].
Ocular
One infant had iridocorneal adhesions and corneal clouding at birth [
Writzl et al 2017].
Conductive hearing loss was present in six of eight individuals; at least one required myringotomy tubes for chronic middle ear effusion [Ehmke et al 2017; Writzl et al 2017; Rodríguez-García et al 2018; Ryu et al 2019; Legué et al 2020; Author, unpublished data].
Dentition
Abdomen/gastrointestinal
Genital
Severe and frequently life-limiting infections have been reported in six individuals [Ehmke et al 2017; Writzl et al 2017; Author, unpublished data]. In two of these individuals, persistent leukocytosis was also present [Writzl et al 2017; Author, unpublished data].
Prognosis
It is unknown whether life span in SLC25A24 Fontaine progeroid syndrome is abnormal. One individual with a molecularly confirmed diagnosis is alive at age 45 years, demonstrating that survival into adulthood is possible [Legué et al 2020].
Additionally, earlier reports of individuals clinically diagnosed with Gorlin-Chaudhry-Moss syndrome and Petty syndrome included individuals in their 30s and 40s [Petty et al 1990, Ippel et al 1992]. Since many adults with disabilities or malformations have not undergone advanced genetic testing, it is likely that adults with this condition are underrecognized and underreported.