Summary
Clinical characteristics.
SLC39A14 deficiency is typically characterized by evidence of delay or loss of motor developmental milestones (e.g., delayed walking, gait disturbance) between ages six months and three years. Early in the disease course, children show axial hypotonia followed by dystonia, spasticity, dysarthria, bulbar dysfunction, and signs of parkinsonism including bradykinesia, hypomimia, and tremor. By the end of the first decade, they develop severe, generalized, pharmaco-resistant dystonia, limb contractures, and scoliosis, and lose independent ambulation. Cognitive impairment appears to be less prominent than motor disability. Some affected children have died in their first decade due to secondary complications such as respiratory infections. One individual with disease onset during the late teens has been reported, suggesting that milder adult presentation can occur.
Diagnosis/testing.
The diagnosis of SLC39A14 deficiency is established in a proband with progressive dystonia-parkinsonism (often combined with other signs such as spasticity and parkinsonian features), characteristic neuroimaging findings, hypermanganesemia, and biallelic pathogenic (or likely pathogenic) variants in SLC39A14 identified on molecular genetic testing.
Management.
Treatment of manifestations: Symptomatic treatment includes physiotherapy and orthopedic management to prevent contractures and maintain ambulation; use of adaptive aids (walker or wheelchair) for gait abnormalities; and use of assistive communication devices. Support by a speech-language pathologist, feeding specialist, and nutritionist to assure adequate nutrition and to reduce the risk of aspiration. When an adequate oral diet can no longer be maintained, gastrostomy tube placement should be considered. Antispasticity medications (baclofen and botulinum toxin) and L-dopa have had limited success. While chelation therapy with intravenous administration of disodium calcium edetate early in the disease course shows promise, additional studies are warranted.
Prevention of primary manifestations: Unknown, but disodium calcium edetate chelation therapy shows promise; additional studies are warranted.
Surveillance: At each visit assess growth, swallowing, and diet to assure adequate nutrition; assess development including ambulation and speech; neurologic examination including scoring of movement disorder severity; consider whole-blood manganese levels and brain MRI as available to assess treatment response and disease progression.
Agents/circumstances to avoid:
- Environmental manganese exposure (i.e., contaminated drinking water, occupational manganese exposure in welding/mining industries, contaminated ephedrone preparations)
- High manganese content of total parenteral nutrition
- Foods very high in manganese including: cloves; saffron; nuts; mussels; dark chocolate; pumpkin, sesame, and sunflower seeds
Evaluation of relatives at risk: Molecular genetic testing for the familial SLC39A14 pathogenic variants of apparently asymptomatic younger sibs of an affected individual allows early identification of sibs who would benefit from prompt initiation of treatment and preventive measures.
Genetic counseling.
SLC39A14 deficiency is inherited in an autosomal recessive manner. Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the SLC39A14 pathogenic variants have been identified in an affected family member, carrier testing of at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
SLC39A14 deficiency should be suspected in probands with typical clinical, neuroimaging, and laboratory findings [Tuschl et al 2016, Garg et al 2022]:
Clinical findings. Infantile or early-childhood onset of the following:
- Delay in acquisition of developmental motor milestones or loss of developmental motor milestones
- Progressive pharmaco-resistant dystonia
- Parkinsonism signs (tremor, bradykinesia, hypomimia)
- Bulbar dysfunction
- Dysarthria
Note: One individual with onset of dystonia during the second decade has been reported, suggesting that milder presentations with juvenile or adult onset may also occur [Namnah et al 2020].
Neuroimaging. Brain MRI findings characteristic of manganese deposition (Figure 1) including T1-weighted hyperintensity of the following:
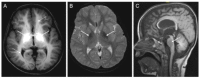
Figure 1
A. Axial T1-weighted image showing the hyperintensity of the globus pallidus (white arrows), and the cerebral white matter (dashed arrows) B. Axial T2-weighted image showing the hypointensity of the globus pallidus (white arrows)
- Globus pallidus and striatum, with thalamic sparing
- Note: Basal ganglia changes on T1-weighted imaging are accompanied by T2-weighted hypointensity.
- White matter including the cerebellum, spinal cord, and dorsal pons, with sparing of the ventral pons
- Anterior pituitary gland
Laboratory findings. Hypermanganesemia. Whole-blood manganese levels are markedly elevated, usually above 1,000 nmol/L (normal reference range <320 nmol/L).
Establishing the Diagnosis
The diagnosis of SLC39A14 deficiency is established in a proband with progressive dystonia (often combined with other signs such as spasticity and parkinsonian features), characteristic neuroimaging findings, hypermanganesemia, and biallelic pathogenic (or likely pathogenic) variants in SLC39A14 identified by molecular genetic testing [Tuschl et al 2016] (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic, and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic SLC39A14 variants of uncertain significance (or of one known SLC39A14 pathogenic variant and one SLC39A14 variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing or a multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires the clinician to determine which gene(s) are likely involved, whereas genomic testing does not. Children with the suggestive clinical, laboratory, and neuroimaging findings could be diagnosed using gene-targeted testing (see Option 1), whereas those with early-onset dystonia-parkinsonism indistinguishable from other inherited disorders with parkinsonism-dystonia are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the clinical, laboratory, and brain MRI findings suggest the diagnosis of SLC39A14 deficiency, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of SLC39A14 is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. If only one or no pathogenic variant is found, gene-targeted deletion/duplication analysis could be considered; however, to date no exon or whole-gene deletions of SLC39A14 have been reported.
- A multigene panel that includes SLC39A14 and other genes of interest (see Differential Diagnosis) may be considered to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the phenotype is indistinguishable from other movement disorders, comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in SLC39A14 Deficiency
Clinical Characteristics
Clinical Description
SLC39A14 deficiency has only recently been identified in 30 individuals from 23 families [Tuschl et al 2016, Anazi et al 2017, Marti-Sanchez et al 2018, Juneja et al 2018, Zeglam et al 2019, Namnah et al 2020, Alhasan et al 2022, Garg et al 2022, Lee & Shin 2022]; therefore, information on the phenotypic spectrum and disease progression is limited.
Onset occurs typically between ages six months and three years. Affected children present with delay or loss of motor developmental milestones (e.g., delayed walking, gait disturbance) [Tuschl et al 2016, Garg et al 2022].
Early in the disease course, children show axial hypotonia followed by dystonia, spasticity, dysarthria, bulbar dysfunction, and signs of parkinsonism including bradykinesia, hypomimia, and tremor.
By the end of the first decade, children develop severe, generalized, pharmaco-resistant dystonia, limb contractures, scoliosis, and loss of independent ambulation.
Although there appears to be relative cognitive sparing (psychometric testing has not been possible), a degree of learning disability is present in all children.
Some affected children die in their first decade due to secondary complications such as respiratory infections.
More recently, one affected individual with a milder phenotype has been described with onset of movement disorder at age 18 years and independent ambulation and survival into late adulthood [Namnah et al 2020].
Neuropathology. The neuropathologic findings in one individual with SLC39A14 deficiency [Tuschl et al 2016] included:
- Extensive gliosis and neuronal loss in the globus pallidus and dentate nucleus;
- Preservation of neurons in the cerebral and cerebellar cortex as well as the caudate, putamen, and thalamus;
- A vacuolated myelinopathy with patchy axonal loss in the cerebral and cerebellar white matter.
Genotype-Phenotype Correlations
No genotype-phenotype correlations are known.
Prevalence
The disease prevalence is not established. To date only 30 individuals with SLC39A14 deficiency from 23 families have been identified. These 23 families are from different ethnic backgrounds and the majority are consanguineous [Tuschl et al 2016, Anazi et al 2017, Marti-Sanchez et al 2018, Juneja et al 2018, Zeglam et al 2019, Namnah et al 2020, Alhasan et al 2022, Garg et al 2022, Lee & Shin 2022].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in SLC39A14.
Differential Diagnosis
Table 2.
Hereditary Disorders of Interest in the Differential Diagnosis of SLC39A14 Deficiency
Additional hereditary disorders in the differential diagnosis of SLC39A14 deficiency include inherited forms of Parkinson disease associated with parkinsonism-dystonia (see Parkinson Disease Overview) and inherited neurodegenerative/metabolic disorders associated with complex dystonia (see Dystonia Overview, Table 4). Both categories of disorders can be distinguished from SLC39A14 deficiency by the absence of features consistent with Mn deposition on brain MRI.
Acquired conditions in the differential diagnosis of SLC39A14 deficiency include acquired hypermanganesemia and acquired hepatocerebral degeneration. Like SLC39A14 deficiency, these disorders of manganese homeostasis are associated with dystonia-parkinsonism, hypermanganesemia, and brain MRI features consistent with manganese deposition.
- Unlike SLC39A14 deficiency, acquired hypermanganesemia often presents with psychiatric symptoms and a history of Mn exposure from environmental sources, parenteral nutrition, or contaminated ephedrone preparations [Mortimer et al 2012, Santos et al 2014, Janocha-Litwin et al 2015].
- Unlike SLC39A14 deficiency, liver disease is the predominant feature in acquired hepatocerebral degeneration and it precedes development of neurologic symptoms [Miletić et al 2014].
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with SLC39A14 deficiency, the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with SLC39A14 Deficiency
Treatment of Manifestations
Symptomatic Treatment
Early initiation of physical therapy and orthopedic management aims to prevent contractures and maintain ambulation. As needed, individuals should be referred for adaptive aids (e.g., a walker or wheelchair for gait abnormalities) and assistive communication devices.
Support by a speech-language pathologist, feeding specialist, and nutritionist is indicated to assure adequate nutrition and to reduce the risk of aspiration. When an adequate oral diet can no longer be maintained, gastrostomy tube placement should be considered. Gastric feeding tube and/or tracheostomy may be required to prevent aspiration pneumonia.
Note that symptomatic treatment with L-dopa and antispasticity medications including benzodiazepines, baclofen, and botulinum toxin has been attempted with limited success. There has been partial but poorly sustained response to trihexyphenidyl at high doses of 20 mg/day and intrathecal baclofen of 1,500-2,000 µg/day in two older sibs reported by Tuschl et al [2016].
Chelation Therapy
There is evidence that disodium calcium edetate, which primarily promotes the urinary excretion of manganese, can improve neurologic symptoms and slow the disease progression [Tuschl et al 2016, Garg et al 2022, Lee & Shin 2022]. Disodium calcium edetate is administered intravenously (20 mg/kg/dose) twice daily for five consecutive days each month.
A female age five years with SLC39A14 deficiency showed improvement of neurologic manifestations with regain of her ability to walk after six months of disodium calcium edetate treatment [Tuschl et al 2016]. In contrast, treatment of a female age 17 years with advanced disease (severe generalized dystonia with prominent oromandibular involvement, contractures, and scoliosis) did not affect disease progression; she continued to deteriorate with worsening tremor and stiffness. Hence, it is likely necessary to initiate chelation treatment early in the disease course.
It is anticipated that chelation therapy will need to be lifelong.
Potential adverse effects of disodium calcium edetate chelation therapy include thrombocytopenia and leukopenia, nephrotoxicity, hepatoxicity, hypocalcemia, and trace metal and vitamin deficiencies [Lamas et al 2012]. Monitoring includes the following:
- Complete blood count
- Assessment of renal function including urinalysis assessed at baseline and monthly thereafter. Monitoring may be extended to every other month once on a stable dose.
- Assessment of liver function
- Measurement of the concentrations of electrolytes, calcium, magnesium, and phosphate
- Measurement of the concentrations of trace metals (manganese, zinc, copper, and selenium)
- Assessment of iron status
Treatment may need to be discontinued if:
- White blood count is <3.5x109/L
- Neutrophil count is <2.0x109/L
- Platelet count is <150x109/L
- >2+ proteinuria is detected on more than one occasion (with no evidence of infection)
The above cut-off values are based on guidelines for D-penicillamine treatment [Chakravarty et al 2008]. Because chelation treatment with disodium calcium edetate may prevent early death and reduce morbidity in SLC39A14 deficiency, lower cut-off values may be acceptable. For each affected individual, the benefits of clinical treatment need to be carefully weighed against the risk of adverse effects.
Prevention of Primary Manifestations
Chelation therapy with disodium calcium edetate may prevent primary disease manifestations in affected sibs who are asymptomatic (see Treatment of Manifestations, Chelation Therapy).
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the following evaluations are recommended.
Table 4.
Recommended Surveillance for Individuals with SLC39A14 Deficiency
Agents/Circumstances to Avoid
The following should be avoided:
- Environmental manganese exposure (i.e., contaminated drinking water, occupational manganese exposure in welding/mining industries, contaminated ephedrone preparations)
- High manganese content of total parenteral nutrition
- Foods very high in manganese including: cloves; saffron; nuts; mussels; dark chocolate; and pumpkin, sesame, and sunflower seeds
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic younger sibs of an affected individual in order to identified as early as possible sibs who would benefit from prompt initiation of treatment and preventive measures (see Agents/Circumstances to Avoid).
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
SLC39A14 deficiency is inherited in an autosomal recessive manner.
Parents of a proband
- The parents of an affected child are presumed to be heterozygous for an SLC39A14 pathogenic variant.
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for an SLC39A14 pathogenic variant and to allow reliable recurrence risk assessment.
- If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, it is possible that one of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017]. If the proband appears to have homozygous pathogenic variants (i.e., the same two pathogenic variants), additional possibilities to consider include:
- A single- or multiexon deletion in the proband that was not detected by sequence analysis and that resulted in the artifactual appearance of homozygosity;
- Uniparental isodisomy for the parental chromosome with the pathogenic variant that resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for an SLC39A14 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. The offspring of an individual with SLC39A14 deficiency are obligate heterozygotes (carriers) for a pathogenic variant in SLC39A14.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an SLC39A14 pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the SLC39A14 pathogenic variants in the family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
- SLC39A14 molecular genetic testing for reproductive partners of known carriers is appropriate, particularly if consanguinity is likely. (The majority of families with SLC39A14 deficiency reported to date have been consanguineous.)
Prenatal Testing and Preimplantation Genetic Testing
Once the SLC39A14 pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- American Parkinson Disease Association (APDA)Phone: 800-223-2732Fax: 718-981-4399Email: apda@apdaparkinson.org
- Dystonia Medical Research FoundationPhone: 312-755-0198; 800-377-DYST (3978)Email: dystonia@dystonia-foundation.org
- Parkinson's FoundationPhone: 800-4PD-INFO (473-4636)Email: contact@parkinson.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
SLC39A14 Deficiency: Genes and Databases
Table B.
OMIM Entries for SLC39A14 Deficiency (View All in OMIM)
Molecular Pathogenesis
SLC39A14 encodes a divalent metal transporter that is required for cellular uptake of manganese [Tuschl et al 2016]. It plays a crucial role as a regulator of manganese homeostasis within the liver and the gut, facilitating biliary manganese excretion and reduced intestinal manganese absorption [Winslow et al 2020].
Biallelic SLC39A14 pathogenic variants are thought to impair hepatic manganese uptake and homeostatic control of intestinal manganese absorption. Subsequently, manganese accumulates in the blood and is deposited in the brain, particularly the globus pallidus, resulting in manganese toxicity and causing progressive dystonia (often combined with other signs such as spasticity and parkinsonian features) [Tuschl et al 2016, Winslow et al 2020].
Mechanism of disease causation. Loss of function
SLC39A14-specific laboratory technical considerations. SLC39A14 comprises nine exons and encodes four transcripts. Two transcripts differ by an alternative 5'UTR (NM_001128431.2 and NM_001135153). Alternative splicing of exon 4 and 9 generates two alternative transcripts (NM_015359.4 and NM_001135154.1).
Chapter Notes
Acknowledgments
This work was supported by grants from Action Medical Research, the Wellcome Trust, Great Ormond Street Hospital Children's Charity, NBIA Disorders Association, Gracious Heart Charity Foundation and Rosetrees Trust, and the GOSH National Institute for Health Research/Biomedical Research Centre.
Revision History
- 8 December 2022 (sw) Comprehensive update posted live
- 25 May 2017 (bp) Review posted live
- 29 November 2016 (kt) Original submission
References
Literature Cited
- Alhasan KA, Alshuaibi W, Hamad MH, Salim S, Jamjoom DZ, Alhashim AH, AlGhamdi MA, Kentab AY, Bashiri FA. Hypermanganesemia with dystonia type 2: a potentially treatable neurodegenerative disorder: a case series in a tertiary university hospital. Children (Basel). 2022;9:1335. [PMC free article: PMC9497897] [PubMed: 36138644]
- Anazi S, Maddirevula S, Faqeih E, Alsedairy H, Alzahrani F, Shamseldin HE, Patel N, Hashem M, Ibrahim N, Abdulwahab F, Ewida N, Alsaif HS, Al Sharif H, Alamoudi W, Kentab A, Bashiri FA, Alnaser M, AlWadei AH, Alfadhel M, Eyaid W, Hashem A, Al Asmari A, Saleh MM, AlSaman A, Alhasan KA, Alsughayir M, Al Shammari M, Mahmoud A, Al-Hassnan ZN, Al-Husain M, Osama Khalil R, Abd El Meguid N, Masri A, Ali R, Ben-Omran T, El Fishway P, Hashish A, Ercan Sencicek A, State M, Alazami AM, Salih MA, Altassan N, Arold ST, Abouelhoda M, Wakil SM, Monies D, Shaheen R, Alkuraya FS. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol Psychiatry. 2017;22:615–24. [PubMed: 27431290]
- Chakravarty K, McDonald H, Pullar T, Taggart A, Chalmers R, Oliver S, Mooney J, Somerville M, Bosworth A, Kennedy T. BSR/BHPR guideline for disease-modifying anti-rheumatic drug (DMARD) therapy in consultation with the British Association of Dermatologists. Rheumatology. 2008;47:924–5. [PubMed: 16940305]
- Garg D, Yoganathan S, Shamim U, Mankad K, Gulati P, Bonifati V, Botre A, Kalane U, Saini AG, Sankhyan N, Srivastava K, Gowda VK, Juneja M, Kamate M, Padmanabha H, Panigrahi D, Pachapure S, Udani V, Kumar A, Pandey S, Thomas M, Danda S, Iqbalahmed SA, Subramanian A, Pemde H, Singh V, Faruq M, Sharma S. Clinical profile and treatment outcomes of hypermanganesemia with dystonia 1 and 2 among 27 Indian children. Mov Disord Clin Pract. 2022;9:886–99. [PMC free article: PMC9547147] [PubMed: 36247901]
- Janocha-Litwin J, Marianska K, Serafinska S, Simon K. Manganese encephalopathy among ephedron abusers. J Neuroimaging. 2015;25:832–5. [PubMed: 25255816]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Juneja M, Shamim U, Joshi A, Mathur A, Uppili B, Sairam S, Ambawat S, Dixit R, Faruq M. A novel mutation in SLC39A14 causing hypermanganesemia associated with infantile onset dystonia. J Gene Med. 2018;20:e3012. [PubMed: 29498153]
- Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, Drisko JA, Lee KL. Design of the trial to assess chelation therapy (TACT). Am Heart J. 2012;163:7–12. [PMC free article: PMC3243954] [PubMed: 22172430]
- Lee JH, Shin JH. Effect of chelation therapy on a Korean patient with brain manganese deposition resulting from a compound heterozygous mutation in the SLC39A14 gene. J Mov Disord. 2022;15:171–4. [PMC free article: PMC9171315] [PubMed: 35306789]
- Marti-Sanchez L, Ortigoza-Escobar JD, Darling A, Villaronga M, Baide H, Molero-Luis M, Batllori M, Vanegas MI, Muchart J, Aquino L, Artuch R, Macaya A, Kurian MA, Dueñas P. Hypermanganesemia due to mutations in SLC39A14: further insights into Mn deposition in the central nervous system. Orphanet J Rare Dis. 2018;13:28. [PMC free article: PMC5791243] [PubMed: 29382362]
- Miletić V, Ozretić D, Relja M. Parkinsonian syndrome and ataxia as a presenting finding of acquired hepatocerebral degeneration. Metab Brain Dis. 2014;29:207–9. [PubMed: 24390157]
- Mortimer JA, Borenstein AR, Nelson LM. Associations of welding and manganese exposure with Parkinson disease: review and meta-analysis. Neurology. 2012;79:1174–80. [PMC free article: PMC3525308] [PubMed: 22965675]
- Namnah M, Bauer M, Mor-Shaked H, Bressman SB, Raymond D, Ozelius LJ, Arkadir D. Benign SLC39A14 course of dystonia-parkinsonism secondary to inherited manganese accumulation. Mov Disord Clin Pract. 2020;7:569–70. [PMC free article: PMC7328422] [PubMed: 32626807]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Santos D, Batoreu C, Mateus L, Marreilha Dos Santos AP, Aschner M. Manganese in human parenteral nutrition: considerations for toxicity and biomonitoring. Neurotoxicology. 2014;43:36–45. [PMC free article: PMC4007395] [PubMed: 24184781]
- Tuschl K, Meyer E, Valdivia LE, Zhao N, Dadswell C, Abdul-Sada A, Hung CY, Simpson MA, Chong WK, Jacques TS, Woltjer RL, Eaton S, Gregory A, Sanford L, Kara E, Houlden H, Cuno SM, Prokisch H, Valletta L, Tiranti V, Younis R, Maher ER, Spencer J, Straatman-Iwanowska A, Gissen P, Selim LA, Pintos-Morell G, Coroleu-Lletget W, Mohammad SS, Yoganathan S, Dale RC, Thomas M, Rihel J, Bodamer OA, Enns CA, Hayflick SJ, Clayton PT, Mills PB, Kurian MA, Wilson SW. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Commun. 2016;7:11601. [PMC free article: PMC4894980] [PubMed: 27231142]
- Winslow JWW, Limesand KH, Zhao N. The functions of ZIP8, ZIP14, and ZnT10 in the regulation of systemic manganese homeostasis. Int J Mol Sci. 2020;21:3304. [PMC free article: PMC7246657] [PubMed: 32392784]
- Zeglam A, Abugrara A, Kabuka M. Autosomal-recessive iron deficiency anemia, dystonia and hypermanganesemia caused by new variant mutation of the manganese transporter gene SLC39A14. Acta Neurol Belg. 2019;119:379–84. [PubMed: 30232769]
Publication Details
Author Information and Affiliations
London, United Kingdom
Portland, Oregon
London, United Kingdom
London, United Kingdom
Portland, Oregon
London, United Kingdom
London, United Kingdom
Publication History
Initial Posting: May 25, 2017; Last Update: December 8, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Tuschl K, Gregory A, Meyer E, et al. SLC39A14 Deficiency. 2017 May 25 [Updated 2022 Dec 8]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
