Summary
NOTE: THIS PUBLICATION HAS BEEN RETIRED. THIS ARCHIVAL VERSION IS FOR HISTORICAL REFERENCE ONLY, AND THE INFORMATION MAY BE OUT OF DATE.
The purpose of this overview is to increase the awareness of clinicians regarding esophageal atresia / tracheoesophageal fistula and its genetic causes and management.
The following are the goals of this overview.
Goal 2.
Review the genetic causes of esophageal atresia / tracheoesophageal fistula.
Goal 3.
Provide an evaluation strategy to identify the genetic cause of esophageal atresia / tracheoesophageal fistula in a proband (when possible).
Goal 4.
Inform genetic counseling of family members of an individual with esophageal atresia / tracheoesophageal fistula.
Goal 5.
Review management of esophageal atresia / tracheoesophageal fistula.
1. Clinical Characteristics of EA/TEF
Clinical Description
Esophageal atresia (EA) is a developmental defect of the upper gastrointestinal tract in which the continuity between the upper and lower esophagus is lost. EA can occur with or without tracheoesophageal fistula (TEF), an abnormal connection between the trachea and the esophagus.
Infants with congenital forms of EA/TEF usually present shortly after birth with copious oral secretions, coughing, gagging, cyanosis, vomiting, and/or respiratory distress.
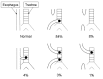
The following five EA/TEF configurations have been described [Clark 1999] (see ):
EA/TEF configurations and their frequencies
EA with a distal TEF (the most common; 84%)
Isolated EA (i.e., without TEF) (8%)
H-type TEF with no EA (4%)
EA with proximal and distal TEF (3%)
EA with a proximal TEF (1%)
A proband with EA/TEF can present in one of the following clinical settings:
Establishing the Diagnosis of EA/TEF
EA/TEF may be suspected prenatally if ultrasound examination reveals polyhydramnios, absence of a fluid-filled stomach, a small abdomen, lower-than-expected fetal weight, and a distended esophageal pouch [Kronemer 2015]. Fetal MRI may be used to confirm the presence of EA/TEF.
EA may be detected postnatally by:
Failure to pass a nasogastric (NG) tube and radiographs that demonstrate coiling of the NG tube in the pouch;
Tracheal compression and deviation on plain chest radiographs;
Absence of a gastric bubble on plain radiographs, which may suggest EA without a TEF or EA with a proximal TEF;
Note: Administration of barium into the esophagus followed by chest radiographs can confirm the diagnosis but is seldom required.
Differential Diagnosis of EA/TEF
Laryngotracheoesophageal cleft. Midline defect between the posterior larynx and trachea and the anterior wall of the esophagus. This entity can present prenatally with polyhydramnios and a small or absent stomach bubble and postnatally with aspiration after feeding. Laryngotracheoesophageal clefts are commonly associated with other anomalies including TEF.
Esophageal webs/rings. Circumferential partial obstruction of the esophageal lumen caused by membranous or diaphragmatic tissue that can be associated with TEF. Many cases are asymptomatic. Symptomatic esophageal webs/rings typically present with recurrent vomiting, dysphagia (solid>liquids), and sometimes aspiration. These symptoms tend to occur later in life than those caused by EA.
Esophageal stricture. A narrowing of the esophageal lumen caused by a variety of intrinsic and extrinsic disease processes. Congenital esophageal strictures typically present after the newborn period in a manner similar to that seen in esophageal webs/rings.
Esophageal diverticulum. A sack or pouch arising from the esophagus. Esophageal diverticula can be present at birth but most often arise and/or become symptomatic in adulthood, presenting with a history of dysphagia, chest pain, vomiting, and sometimes aspiration pneumonia.
Tubular esophageal
duplications. Tubular channels that lie parallel to the esophagus and often connect to the main esophageal lumen or the stomach. These lesions are often asymptomatic and are most commonly identified serendipitously at autopsy. Symptoms typically result from inflammation and/or distention secondary to food entrapment and typically involve intermittent dysphagia.
Congenital short esophagus. An abnormally short esophagus accompanied by an intrathoracic location of part of the stomach. Symptoms are often present at birth and include gastroesophageal reflux and vomiting.
Tracheal agenesis/atresia. Lack of communication between the larynx and the alveoli of the lungs. This can be associated with other anomalies including TEF. Prenatal findings can include hyperechoic lungs, flattened diaphragms, oligo- or polyhydramnios, and large breathing movements. Postnatal symptoms include severe respiratory distress, cyanosis, absent cry, and failure to ventilate despite tracheal intubation.
2. Genetic Causes of EA/TEF
The genetic causes of esophageal atresia / tracheoesophageal fistula (EA/TEF) include syndromes that result from mutation of a single gene (see Table 1) or chromosome anomalies.
Table 1.
Esophageal Atresia / Tracheoesophageal Fistula: Syndromic Causes and Distinguishing Clinical Features
View in own window
| Syndrome 1 | Gene | MOI | Distinguishing Clinical Findings |
|---|
|
Anophthalmia-esophageal-genital syndrome
|
SOX2
| AR |
|
|
CHARGE syndrome
|
CHD7
| AR | Coloboma of the eye Cardiac anomalies Choanal atresia Intellectual disability Growth restriction Genital anomalies Ear anomalies Hearing loss EA/TEF
|
|
Feingold syndrome
|
MYCN
| AR |
|
|
Fanconi anemia
| ≥20 genes | AR
AD | Bone marrow failure Malignancies Short stature Abnormal skin pigmentation Radial ray defects Eye anomalies Renal anomalies Cardiac defects Abnormal ears Central nervous system anomalies Hearing loss Developmental delay Gastrointestinal anomalies incl EA/TEF
|
| VACTERL-H (OMIM 300515) |
FANCB
| XL |
|
- 1.
EA/TEF has also been reported in individuals with Opitz G/BBB and Pallister-Hall syndrome, although it is not a major feature in these disorders.
Chromosomal Causes of EA/TEF
Chromosome anomalies have been reported in approximately 6%-10% of individuals with EA/TEF. EA/TEF is found in the following aneuploidy syndromes [Felix et al 2007, Brosens et al 2014a]:
Copy number variant (CNV) analysis in individuals with both isolated and non-isolated EA/TEF can reveal rare gene-containing CNVs, a subset of which can be shown to have arisen de novo [Brosens et al 2014b].
Recurrent duplications of 3p25-pter and 5q34-qter suggest that these regions may harbor one or more genes in which overexpression causes or predisposes to the development of EA/TEF [Felix et al 2007].
Recurrent deletions of 2q37.2-qter, 4q35-qter, 5p15-pter, 6q13-q15, 14q32.3-qter, and 17q22-q23 suggest that haploinsufficiency or decreased expression of one or more genes in these regions may cause or predispose to the development of EA/TEF [Felix et al 2007].
Recurrent deletions of 13q34-qter and 22q11 are also associated with EA/TEF, but typically with findings consistent with VACTERL association [Walsh et al 2001, Felix et al 2007].
3. Evaluation Strategies to Identify the Genetic Cause of EA/TEF in a Proband
Establishing a specific genetic cause of EA/TEF:
Can aid in discussions of prognosis (which are beyond the scope of this GeneReview) and genetic counseling;
Usually involves a medical history, physical examination, laboratory testing, family history, and genomic/genetic testing.
Medical history. A history of neurodevelopmental defects, other structural birth defects, abnormal growth patterns, bone marrow failure, malignancies, and hearing loss may provide evidence in support of a specific cause of EA/TEF (see Table 1).
Physical examination. The identification of other structural birth defects (e.g., anal anomalies, limb defect) and/or abnormal growth patterns may provide evidence in support of a specific cause of EA/TEF (see Table 1).
Family history. A three-generation family history should be taken, with attention to relatives with manifestations of EA/TEF and documentation of relevant findings through direct examination or review of medical records, including results of molecular genetic testing.
Imaging studies may identify additional anomalies of the skeleton, heart, and/or kidneys.
Cytogenetic testing. If aneuploidy is suspected in an individual with EA/TEF, a G-banded karyotype can be performed (see Chromosomal Causes of EA/TEF).
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing or multigene panel), chromosomal microarray analysis [CMA], and comprehensive genomic testing (exome sequencing, genome sequencing, or exome array). Gene-targeted testing requires the clinician to hypothesize which gene(s) are likely involved, whereas genomic testing does not.
Serial single-gene testing can be considered if clinical findings and/or family history indicate that pathogenic variants in a particular gene are most likely causative (see
Table1).
A multigene panel that includes some or all of the genes listed in the
Table 1 is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see
Table 1).
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Chromosomal microarray analysis (CMA) using oligonucleotide or SNP arrays to detect genome-wide large deletions/duplications that cannot be detected by sequence analysis may be considered in individuals with distinguishing phenotypic features suggestive of a contiguous gene deletion (see
Chromosomal Causes of EA/TEF).
Comprehensive
genomic testing (which does not require the clinician to determine which gene[s] are likely involved) may be considered. Exome sequencing is most commonly used; genome sequencing is also possible.
Exome array (when clinically available) may be considered if exome sequencing is not diagnostic.
For an introduction to comprehensive genomic testing click
here. More detailed information for clinicians ordering genomic testing can be found
here.
4. Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Esophageal atresia / tracheoesophageal fistula (EA/TEF) can occur as an isolated finding, as part of a genetic syndrome, or as part of a non-isolated (but not syndromic) set of findings. Most individuals with EA/TEF represent simplex cases (i.e., the only affected member of the family). A small subset of families are multiplex (i.e., ≥2 relatives have EA/TEF).
Nonsyndromic EA/TEF is generally considered to be inherited in a multifactorial manner.
If an affected individual is found to have an inherited or de novo chromosome abnormality, or a specific syndrome or association with EA/TEF, genetic counseling for that condition is indicated.
Risk to Family Members
Table 2.
View in own window
| EA/TEF Classification | Recurrence Risk |
|---|
| To sibs of a proband | To offspring of a proband |
|---|
| Isolated w/o a clear etiology 1 | Empiric risk: 1% 2 | ~2%-4% 2, 3, 4 |
| Non-isolated & part of a known genetic syndrome | Depends on:
| Depends on MOI assoc'd w/the genetic syndrome |
| Non-isolated & not part of a known genetic syndrome 5 |
| ~2%-4% 2, 3 |
EA/TEF = esophageal atresia / tracheoesophageal fistula; MOI = mode of inheritance
- 1.
Nonsyndromic EA/TEF is generally considered to be inherited in a multifactorial manner. The twin concordance rate for EA/TEF is also low (~2.5%) [Robert et al 1993].
- 2.
- 3.
Recurrence risk for EA/TEF and/or malformations in the VACTERL association.
- 4.
Since these estimates were generated by studying the offspring of individuals with both isolated and non-isolated EA/TEF, the risk to offspring in isolated cases may be lower [Shaw-Smith 2006].
- 5.
Recurrence risk counseling for individuals with non-isolated EA/TEF – in whom additional anomalies have been identified and for whom a specific genetic disorder is not recognized – is problematic:
• Some cases of non-isolated EA/TEF are probably caused by de novo autosomal dominant mutations, and therefore, pose a low recurrence risk to the sibs of the proband.
• Some are probably unrecognized or private autosomal recessive conditions.
• Some may be multifactorial disorders with a low recurrence risk.
• Non-genetic causes including stochastic events, epigenetic modifications, or teratogenic/environmental exposures are possible as well.
- 6.
Counseling in this setting should be as for other multiple congenital anomaly disorders of unknown etiology.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. If the pathogenic variant(s) have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Ultrasound examination
Prenatal ultrasound examination of a fetus affected by EA/TEF may reveal an absent or small fetal stomach bubble in combination with maternal polyhydramnios [
Houben & Curry 2008]. The diagnostic accuracy is increased if an anechoic area is present in the middle of the fetal neck, as it can help differentiate EA from diseases with possible swallowing impairments [
Kronemer 2015]. In one study of 87 fetuses with a small (n = 53) or absent (n = 34) stomach bubble, the positive predictive value of an absent stomach bubble and polyhydramnios was 56% and the sensitivity of prenatal sonography in the diagnosis of EA was 42% based on the outcome of all fetuses scanned during the same period [
Stringer et al 1995]. The positive predictive value is likely to be even lower among fetuses with multiple anomalies [
Choudhry et al 2007].
The presence of a dilated blind-ending esophageal pouch on a sonogram is suggestive of EA. It is the most reliable sonographic sign indicative of EA and has been seen in cases with and without TEF [
Kronemer 2015].
Polyhydramnios can be seen in a wide variety of disorders and alone is a poor indicator of EA: only one in 12 fetuses with polyhydramnios has EA [
Kronemer 2015].
When EA/TEF is suspected on routine prenatal ultrasound examination:
A high-resolution ultrasound examination is indicated;
Fetal MRI should be considered to help confirm the diagnosis of EA/TEF and to evaluate for additional structural anomalies;
Chromosome analysis, an array-based copy number detection assay, and/or molecular testing using fetal cells/DNA should be considered.
All fetuses suspected of having EA/TEF should be evaluated for the presence of additional major malformations that could be part of an underlying syndrome because such malformations and/or the underlying diagnosis may significantly affect the prognosis. Involvement of a clinical geneticist in the evaluation of these families can be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
National Library of Medicine Genetics Home Reference
TOFS
St. George's Centre
91 Victoria Road
Netherfield Nottingham NG4 2NN
United Kingdom
Phone: +44 (0)115 961 3092
Fax: +44 (0)115 961 3097
Email: info@tofs.org.uk
Medline Plus
5. Management of EA/TEF
Evaluations Following Initial Diagnosis
After an individual has been diagnosed with esophageal atresia / tracheoesophageal fistula (EA/TEF), the following evaluations should be considered as a means of identifying associated anomalies:
Treatment of Manifestations
Initial postnatal intervention, aimed at minimizing the risk of aspiration pneumonia, typically includes the elimination of oral feeds, placement of a suction catheter to allow continuous drainage of secretions, and elevation of the head of the bed to minimize reflux [Clark 1999]. Intravenous (IV) glucose and fluids should be provided; supplemental oxygen should be administered as needed. When intubation cannot be avoided, a possible complication is the collection of air in the stomach, which in severe cases can be removed only by gastrostomy.
Surgical repair consists of closure of the TEF and anastomosis of the esophageal segments [Sharma 2017]. Surgical repair may need to be delayed in infants with low birth weight, pneumonia, or other major congenital anomalies. When surgical repair is delayed, infants may be treated with parenteral nutrition, gastrostomy tube placement, and upper pouch suctioning until they become surgical candidates.
The most common complications after surgical repair include leakage at the site of the anastomosis, recurrent fistula, structure formation, and gastroesophageal reflux [Clark 1999, Kronemer 2015, Sharma 2017].
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Chapter Notes
Author Notes
Baylor College of Medicine Molecular and Human Genetics web page
Revision History
20 April 2023 (ma) Chapter retired: chapter does not reflect current use of genetic testing
20 September 2018 (sw) Comprehensive update posted live
12 June 2014 (me) Comprehensive update posted live
12 March 2009 (me) Review posted live
14 October 2008 (das) Original submission
References
Literature Cited
Brosens E, de Jong EM, Barakat TS, Eussen BH, D'Haene B, De Baere E, Verdin H, Poddighe PJ, Galjaard RJ, Gribnau J, Brooks AS, Tibboel D, de Klein A. Structural and numerical changes of chromosome X in patients with esophageal atresia.
Eur J Hum Genet. 2014a;22:1077–84. [
PMC free article: PMC4135408] [
PubMed: 24398799]
Brosens E, Ploeg M, van Bever Y, Koopmans AE, Ijsselstijn H, Rottier RJ, Wijnen R, Tibboel D, de Klein A. Clinical and etiological heterogeneity in patients with tracheo-esophageal malformations and associated anomalies.
Eur J MED Genet. 2014b;57:440–52. [
PubMed: 24931924]
Castori M, Rinaldi R, Capocaccia P, Roggini M, Grammatico P. VACTERL association and maternal diabetes: a possible causal relationship?
Birth Defects Res A Clin Mol Teratol. 2008;82:169–72. [
PubMed: 18181216]
Choudhry M, Boyd PA, Chamberlain PF, Lakhoo K. Prenatal diagnosis of tracheo-oesophageal fistula and oesophageal atresia.
Prenat Diagn. 2007;27:608–10. [
PubMed: 17457956]
Clark DC. Esophageal atresia and tracheoesophageal fistula.
Am Fam Physician. 1999;59:910–6, 919-20. [
PubMed: 10068713]
Felix JF, Tibboel D, de Klein A. Chromosomal anomalies in the aetiology of oesophageal atresia and tracheo-oesophageal fistula.
Eur J Med Genet. 2007;50:163–75. [
PubMed: 17336605]
Houben CH, Curry JI. Current status of prenatal diagnosis, operative management and outcome of esophageal atresia/tracheo-esophageal fistula.
Prenat Diagn. 2008;28:667–75. [
PubMed: 18302317]
Kronemer KA. Imaging in esophageal atresia and tracheoesophageal fistula. Medscape. Available
online (registration required). 2015. Accessed 4-11-23.
Robert E, Mutchinick O, Mastroiacovo P, Knudsen LB, Daltveit AK, Castilla EE, Lancaster P, Källén B, Cocchi G. An international collaborative study of the epidemiology of esophageal atresia or stenosis.
Reprod Toxicol. 1993;7:405–21. [
PubMed: 8274816]
Sharma S. Tracheoesophageal fistula. Medscape. Available
online (registration required). 2017. Accessed 4-11-23.
Stringer MD, McKenna KM, Goldstein RB, Filly RA, Adzick NS, Harrison MR. Prenatal diagnosis of esophageal atresia.
J Pediatr Surg. 1995;30:1258–63. [
PubMed: 8523220]
Walsh LE, Vance GH, Weaver DD. Distal 13q deletion syndrome and the VACTERL association: case report, literature review, and possible implications.
Am J Med Genet. 2001;98:137–44. [
PubMed: 11223849]