Summary
Clinical characteristics.
The clinical features of TNXB-related classical-like Ehlers-Danlos syndrome (clEDS) strongly resemble those seen in classic EDS (cEDS). Affected individuals have generalized joint hypermobility, hyperextensible skin, and easy bruising, but do not have atrophic scarring, as is seen in cEDS. There are also several other distinguishing clinical findings including anomalies of feet and hands, edema in the legs in the absence of cardiac failure, mild proximal and distal muscle weakness, and axonal polyneuropathy. Vaginal, uterine, and/or rectal prolapse can also occur. Tissue fragility with resulting rupture of the trachea, esophagus, and small and large bowel has been reported. Vascular fragility causing a major event occurs in a minority of individuals. Significant variability in the severity of musculoskeletal symptoms and their effect on day-to-day function between unrelated affected individuals as well as among affected individuals in the same family has been reported. Fatigue has been reported in more than half of affected individuals. The severity of symptoms in middle-aged individuals can range from joint hypermobility without complications to being wheelchair-bound as a result of severe and painful foot deformities and fatigue.
Diagnosis/testing.
The diagnosis of TNXB-related clEDS is established in a proband with suggestive clinical findings and biallelic pathogenic variants in TNXB identified by molecular genetic testing.
Management.
Treatment of manifestations: Non-weight-bearing exercise, physical therapy, and careful selection of analgesic medication to address joint pain; if pain is severe or debilitating, consider referral to a pain management specialist or clinic. The use of opioid medication should be avoided for chronic pain, as this does not lead to long-term pain relief and has the potential for addiction issues. Long-term chronic pain may result in the need for mental health services. Prompt assessment and management in a tertiary center is required for bowel rupture, arterial rupture, or organ rupture. Ascorbic acid (vitamin C) may reduce easy bruising but has no effect on the key characteristics of skin hyperextensibility and joint hypermobility. DDAVP® (deamino-delta-D-arginine vasopressin) may also be useful to normalize bleeding time in those with easy bruising. Standard treatment is applicable for joint dislocations, subjective muscle weakness, and cardiac abnormalities.
Prevention of primary manifestations: Maintenance of a health body weight; low threshold for referral to gastroenterologist for evaluation of gastrointestinal symptoms; special attention during general anesthesia in order to provide adequate positioning and support as well as being aware of tissue fragility, which has been reported after intubation. Avoid invasive procedures unless absolutely medically necessary; consider carrying medical information or wearing jewelry denoting an increased risk of tissue fragility.
Surveillance: Routine follow up ideally with rheumatologist, pain management clinic, and/or specialized EDS services, if available.
Agents/circumstances to avoid: Sports with heavy joint strain, as well as contact sports; invasive procedures unless they are absolutely medically necessary; acetylsalicylate (aspirin) and long-term use of NSAIDS; use of opioid medication for chronic pain.
Pregnancy management: It is important that the obstetrician and midwives are made aware of the diagnosis of clEDS during a pregnancy. Reported pregnancy and postpartum issues in affected women include miscarriage, premature rupture of membranes, post- or peripartum hemorrhage, and prolapse of the rectum, vagina, and/or uterus. Specialist delivery is strongly advised in view of reported trachea rupture during intubation and esophagus rupture after insertion of a transesophageal ultrasound probe.
Genetic counseling.
TNXB-related clED is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a TNXB pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being heterozygous, and a 25% chance of inheriting neither of the familial pathogenic variants. Once the TNXB pathogenic variants have been identified in an affected family member, carrier testing and prenatal/preimplantation genetic testing are theoretically possible.
Diagnosis
Minimum suggestive clinical diagnostic criteria for TNXB-related classical-like Ehlers-Danlos syndrome (clEDS) were published in the 2017 revised Ehlers-Danlos syndrome nosology [Malfait et al 2017] (full text), and include all three major criteria AND a family history compatible with autosomal recessive inheritance (see Suggestive Findings).
Suggestive Findings
TNXB-related clEDS should be suspected in individuals with a combination of the following major, minor, and family history criteria [Malfait et al 2017].
Major criteria
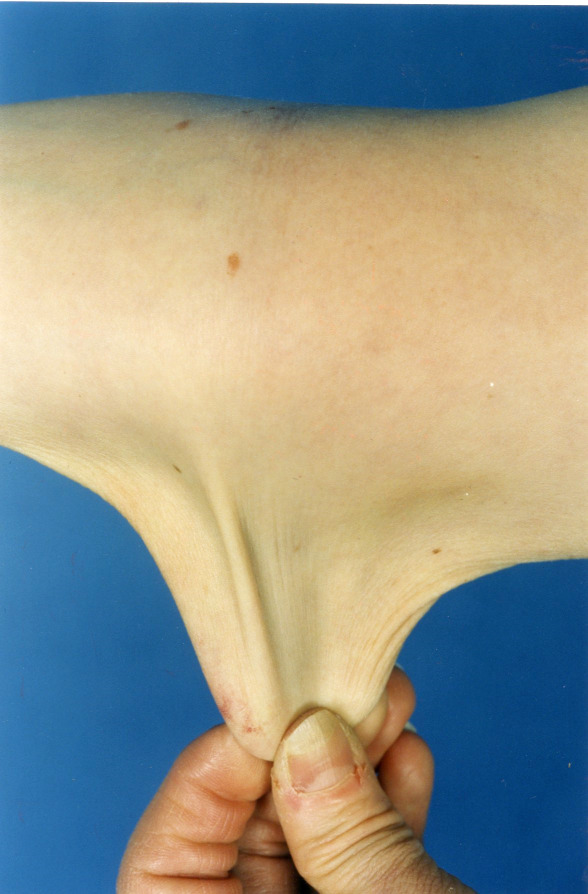
- Skin hyperextensibility with velvety skin texture and absence of atrophic scarring (See Figure 1.)
- Hyperextensibility can be objectively measured by pinching the cutaneous and subcutaneous layers of skin located in the middle of the volar surface of the nondominant forearm and stretching it to at least 1.5 cm or at least 1 cm on the volar surface of the hand (palm).
- For the neck, elbow, and knees, the stretched measurement should be at least 3 cm.
- Generalized joint hypermobility with or without recurrent dislocations (most commonly shoulder and ankle)
- Generalized joint hypermobility is typically measured using a Beighton score (see Classic Ehlers-Danlos Syndrome, Table 1; Malfait et al [2017]).
- A score of ≥5 at some point in life is considered positive.
- Easy or spontaneous bruising of the skin
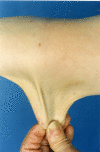
Figure 1.
Hyperextensible skin at the elbow
Minor criteria
- Hand anomalies including:
- Acrogeric hands (characterized by thinning and wrinkling of the skin) with excessive skin
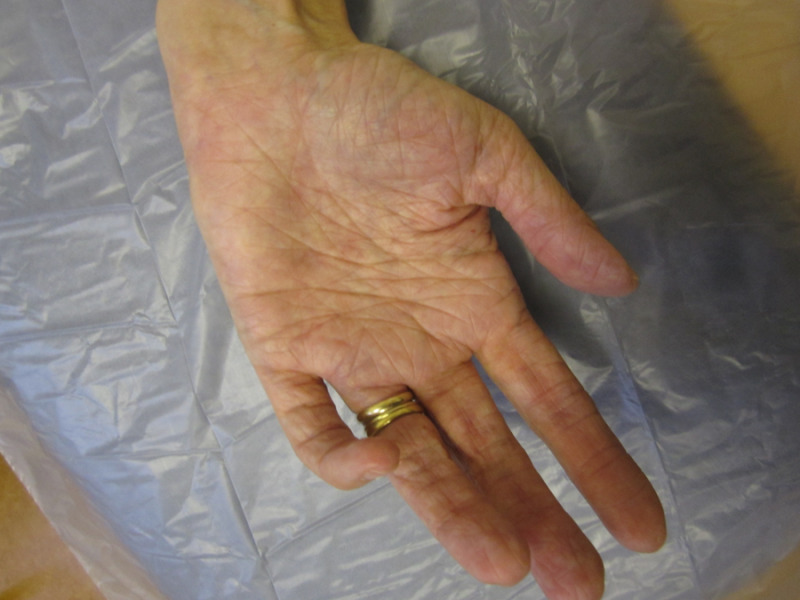
- Mallet finger(s) (See Figure 2.)
- Clinodactyly
- Brachydactyly
- Atrophy of the muscles in the hands and feet
- Foot anomalies including:
- Broad/plump forefoot
- Brachydactyly with excessive skin
- Pes planus
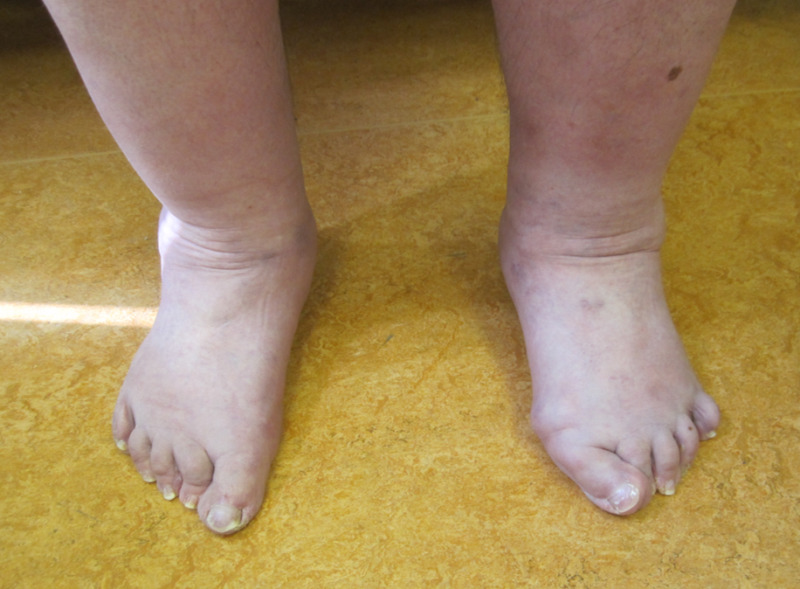
- Hallux valgus (See Figure 3.)
- Piezogenic papules
- Edema in the legs in the absence of cardiac failure
- Mild proximal and distal muscle weakness
- Axonal polyneuropathy
- Vaginal, uterus, and/or rectal prolapse
- Predisposition to tissue fragility, particularly of the gastrointestinal tract (a feature usually suggestive of vascular Ehlers-Danlos syndrome; see Differential Diagnosis.)Note: This finding was proposed as an additional important feature of TNXB-related clEDS by Green et al [2020] but was not included in the 2017 revised Ehlers-Danlos syndrome nosology [Malfait et al 2017].

Figure 2.
Mallet finger (digit 5)
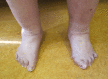
Figure 3.
Flat, broad feet with left hallux valgus and bilateral hammer toes of digits 2-4
Family history is compatible with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis [Malfait et al 2017].
Establishing the Diagnosis
The diagnosis of TNXB-related clEDS is established in a proband with suggestive clinical findings and biallelic pathogenic (or likely pathogenic) variants in TNXB identified by molecular genetic testing (see Table 1). See Molecular Genetics for more information about the technical challenges related to genetic testing for this gene.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic TNXB variants of uncertain significance (or identification of one known TNXB pathogenic variant and one TNXB variant of uncertain significance) does not establish or rule out a diagnosis of this disorder.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype. Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of TNXB-related clEDS has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and family history findings suggest the diagnosis of TNXB-related clEDS, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis * of TNXB is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.* See Molecular Genetics for technical considerations pertaining to sequence analysis for this gene due to the TNXA pseudogene.
- An Ehlers-Danlos syndrome or connective tissue disorders multigene panel that includes TNXB and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) TNXB exome analysis can be complicated by the TNXA pseudogene; see Molecular Genetics. (2) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (3) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (4) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (5) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. Copy number variant analysis should be performed to detect deletions/duplications, which have been reported in affected individuals [Demirdas et al 2017].
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by connective tissue findings, comprehensive genomic testing may be considered.
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in TNXB-Related Classical-Like Ehlers-Danlos Syndrome
Other laboratory findings. In the past, detection of tenascin-X in serum was performed and reported to be absent in individuals with TNXB-related clEDS [Schalkwijk et al 2001]. However, no diagnostic laboratory currently offers this test.
Clinical Characteristics
Clinical Description
TNXB-related classical-like Ehlers-Danlos syndrome (clEDS) was first reported in 1997 [Burch et al 1997]. It was noted that the clinical features of these individuals strongly resembled classic EDS (cEDS) because of hyperextensible skin and generalized hypermobility, with two key differences: (1) absence of atrophic scarring and (2) autosomal recessive inheritance. Due to the clinical resemblance with cEDS, updated nosology has renamed the condition classical-like EDS (clEDS) [Malfait et al 2017]. Since the first publications, other features have been reported in individuals with clEDS that are more specific to clEDS [Brady et al 2017, Demirdas et al 2017]. These are reflected in the minor criteria for clEDS and include broad feet and hands, brachydactyly, edema in the legs in the absence of cardiac failure, and predisposition to tissue fragility, particularly of the gastrointestinal tract [Green et al 2020].
To date, 56 individuals from 44 families have been identified with TNXB-related clEDS [Burch et al 1997, Schalkwijk et al 2001, Lindor & Bristow 2005, Voermans et al 2007, Voermans et al 2009, Besselink-Lobanova et al 2010, O'Connell et al 2010, Hendriks et al 2012, Pénisson-Besnier et al 2013, Sakiyama et al 2015, Demirdas et al 2017, Micale et al 2019, Rymen et al 2019, Brisset et al 2020, Green et al 2020, Watanabe et al 2021]. The following description of the phenotypic features associated with this condition is based on these reports. It is important to note that the majority of affected individuals were diagnosed in adulthood.
Table 2.
TNXB-Related Classical-Like Ehlers-Danlos Syndrome: Frequency of Select Features
Skin. People with TNXB-related clEDS invariably have hyperextensible skin.
- Although absence of atrophic scarring is one of the features that distinguishes TNXB-related clEDS from cEDS, mild atrophic scarring (not including cigarette paper scarring and hemosiderosis) has been reported in seven affected individuals.
- Easy bruising is reported in the majority of affected individuals. In one individual a suspicion of nonaccidental injury had been raised due to excessive bruising.
- Hematomas are frequently encountered.
Musculoskeletal. Generalized joint hypermobility is always present in affected individuals, and many have recurrent joint (sub)luxations. Foot deformities (listed in Suggestive Findings) are present in the majority; hand abnormalities (acrogeric hands, mallet finger[s], clinodactyly, and brachydactyly) are less frequently reported [Demirdas et al 2017, Green et al 2020].
- Significant variability between both unrelated and related affected individuals in the severity of musculoskeletal symptoms and their effect on day-to-day function has been reported.
- The severity of symptoms in middle-aged individuals can range from joint hypermobility without complications to being wheelchair-bound due to severe and painful foot deformities, joint dislocations, and fatigue [Green et al 2020].
- More than half of affected individuals reported fatigue as an important feature (e.g., Demirdas et al [2017]: 14/17 individuals; Green et al [2020]: 14/20 individuals).
- Edema of the ankles and/or feet has been described in 14 of 56 individuals and could not be attributed to a cardiac etiology.
Cardiovascular
- Vascular fragility has been reported in 15 of 56 (27%) affected individuals, with three of 56 (5%) experiencing major medical events due to vascular fragility.
- Ten individuals experienced frequent subconjunctival hemorrhages.
- Rupture of a right brachial vein was reported in a woman age 26 years [Micale et al 2019].
- A man age 58 years had a thoraco-abdominal aortic aneurysm and aneurysm of both the common iliac artery and superior mesenteric artery [Demirdas et al 2017].
- An affected individual who died in his sixth decade due to a bowel rupture had aneurysmal abdominal arteries on postmortem examination [Demirdas et al 2017].
- One individual required surgery for two separate incidences of spontaneous compartment syndrome in his right and left arm at age 30 and 31 years, respectively [Green et al 2020].
- One individual developed a spontaneous left-calf hematoma that had to be drained surgically [Green et al 2020].This individual also developed a right-arm cephalic vein thrombosis and pulmonary embolism during admission for adrenal crisis.She was subsequently started on anticoagulant therapy and shortly after required a hospital admission for spontaneous subcutaneous hematoma of the lower half of the body, causing acute anemia and requiring blood transfusion.
- Valvular anomalies. Mild valvular abnormalities, often involving the mitral valve, have been reported in nine of 56 (16%) of affected individuals.
- Cardiomyopathy was detected in three of 56 (5%) individuals (postpartum, dilated, and unspecified, respectively). It remains unclear if this represents rare co-occurrences of cardiomyopathy with TNXB-related clEDS or if cardiomyopathy is a rare feature of TNXB-related clEDS.
Neuromuscular
- Subjective muscle weakness has been reported in about one third of affected individuals. Based partially on physiologic studies in affected individuals, a dose-effect relation of TNX levels and degree of neuromuscular involvement has been suggested [Castori & Voermans 2014].The most elaborate study included ten individuals with TNXB-related clEDS among a group of 40 individuals with EDS of varying types [Voermans et al 2009].Those with TNXB-related clEDS generally had moderate neuromuscular complaints, mild reduced sensation, muscle weakness, and functional impairment on physical examination.
- Moderate polyneuropathy and mild abnormal motor unit action potentials were seen on clinical neurophysiologic studies.
- Muscle ultrasound demonstrated increased echo intensity.
- Muscle biopsy showed mild myopathic changes in some affected individuals.
- It has been hypothesized that neuropathy may be linked to increased vulnerability of peripheral nerves to stretching/pressure due to TNX deficiency [Castori & Voermans 2014].
- Atrophy of the muscles in the hands and feet has been reported in 4% of affected individuals, although this feature was not assessed by Green et al [2020] in their cohort of 20 affected individuals. It is unclear whether this is a characteristic feature of TNXB-related clEDS.
Gastrointestinal
- Rupture. Nine affected individuals have been reported with gastrointestinal fragility leading to a total of 14 gastrointestinal events. Four of these individuals had more than one gastrointestinal event in different locations, and four had a gastrointestinal event during an invasive procedure. One of these latter four had three events, two of which occurred after an invasive procedure. Age at which first gastrointestinal complications occurred varied from 36 years to 59 years. These findings imply a degree of tissue fragility with complications resulting from invasive procedures. The nine affected individuals and their respective events were as follows:
- A man age 36 years with a perforation of a colonic diverticulum, who developed multiple abscesses requiring partial colectomy, which was complicated by a second small-bowel perforation [Lindor & Bristow 2005]
- A man age 57 years who experienced an esophageal rupture possibly resulting from an ultrasound probe [Hendriks et al 2012]
- A woman age 45 years who had a diverticular perforation of the sigmoid colon and duodenal perforation after ileus tube insertion [Sakiyama et al 2015]
- An individual age 48 years who had a bowel perforation as a result of diverticulitis [Demirdas et al 2017]
- An individual age 38 years with a colonic perforation during a colonoscopy [Brisset et al 2020]
- A woman age 42 years who had a spontaneous perforation of the small bowel for which an intestinal specimen was reported to be "very fragile" [Rymen et al 2019]
- Spontaneous transverse colon perforation at age 51 years followed by a second perforation of the small bowel three days postoperative [Green et al 2020]
- Jejunal perforation at age 40 years [Green et al 2020]
- Esophageal perforation at age 55 years during a gastroscopy, followed by a spontaneous small bowel perforation at age 56 years, and one small bowel perforation after a nasojejunal barium study at age 59 years [Green et al 2020]
Note: Demirdas et al [2017] reported death as a result of infection following bowel perforation of an individual in the sixth decade as unpublished data (not included in the cross-sectional analysis of 17 individuals represented in the publication). - Diverticular disease has been noted in ten of 56 affected individuals. It is hypothesized that affected individuals may be more prone to structural defects along the walls of the gastrointestinal tract, which can predispose to diffuse diverticulosis, diverticulitis, and resulting bowel perforation as well as to perforation during invasive procedures and spontaneous perforation [Green et al 2020].
- Gastrointestinal bleeding has been reported in three of 56 affected individuals. It was not specified whether this occurred spontaneously or due to another gastrointestinal issue.
Other tissue fragility. Tracheal rupture possibly due to intubation has been reported in a woman age 41 years [Besselink-Lobanova et al 2010]. A woman age 47 years had extensive surgical emphysema within the subcutaneous tissues of her face and computed tomography (CT) scans of the sinuses revealed a defect of the left nasal cartilages anteriorly, allowing air to track into the soft tissues. It was felt that vigorous nose blowing had most likely been the cause of the emphysema. This may also point to tissue fragility in individuals with TNXB-related clEDS [Green et al 2020].
Organ prolapse. As in other types of EDS, vaginal/uterus/rectal prolapse are more frequently encountered and have been reported in 21% of individuals with TNXB-related clEDS.
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been identified.
Nomenclature
Outdated terms for TNXB-related clEDS include tenascin-X deficient type of Ehlers-Danlos syndrome and tenascin-X deficiency.
Prevalence
The prevalence of TNXB-related clEDS is unknown; 56 individuals from 44 families have been described in the literature. Two additional affected individuals were referenced by Demirdas et al [2017] as "unpublished data."
Genetically Related (Allelic) Disorders
CAH-X syndrome. In 20%-30% of individuals with severe salt-wasting 21-hydroxylase-deficient congenital adrenal hyperplasia (21-OHD CAH), deletions of CYP21A2 are identified as a result of unequal crossover during meiosis. These deletions are categorized into two subtypes: CAH and CAH-X. There are three distinct CAH-X chimeras that impair both CYP21A2 and TNXB. Biallelic CAH-X deletions lead to 21-OHD CAH with TNXB-related clEDS. In individuals with a monoallelic CAH-X deletion, joint hypermobility, pes planus, and hernias have been observed as well as congenital heart defects and other features [Miller & Merke 2018]. However, further studies in more individuals are needed to establish a causal relationship.
Vesicoureteral reflux (VUR) 8 (OMIM 615963). In a five-generation family with various members affected with VUR, Gbadegesin et al [2013] identified a heterozygous missense variant in TNXB. Subsequently, in 11 other families, TNXB was analyzed and a maternally inherited missense variant was identified in one affected individual; a voiding cystourethrogram was not performed on the mother to enable detection of vesicoureteral reflux. Currently, there is not enough evidence to support a causal relationship between TNXB variant(s) and VUR.
Differential Diagnosis
Table 3.
Genes and Disorders in the Differential Diagnosis of TNXB-Related Classic-Like Ehlers-Danlos Syndrome
Hypermobile Ehlers-Danlos syndrome (hEDS) should also be considered in the differential diagnosis of TNXB-related clEDS, as hEDS can be associated with mild atrophic scarring, generalized joint hypermobility with or without joint (sub)luxations, and easy bruising. However, the absence of clearly hyperextensible skin in hEDS distinguishes this disorder from TNXB-related clEDS. The diagnosis of hEDS is based entirely on clinical evaluation and family history. The molecular basis of hEDS is unknown.
Management
No consensus clinical practice guidelines for TNXB-related classical-like Ehlers-Danlos syndrome (clEDS) have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with TNXB-related clEDS, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with TNXB-Related Classical-Like Ehlers-Danlos Syndrome
Treatment of Manifestations
Table 5.
Treatment of Manifestations in Individuals with TNXB-Related Classical-Like Ehlers-Danlos Syndrome
Prevention of Primary Manifestations
For recommendations on prevention of primary manifestations of joint laxity and dislocations as well as joint pain, see Hypermobile Ehlers-Danlos Syndrome, Prevention of Primary Manifestations.
Table 6.
Prevention of Primary Manifestations in Individuals with TNXB-Related Classical-Like Ehlers-Danlos Syndrome
Surveillance
Table 7.
Recommended Surveillance for Individuals with TNXB-Related Classical-Like Ehlers-Danlos Syndrome
Agents/Circumstances to Avoid
The following should be avoided:
- Sports with heavy joint strain (e.g., contact sports, fighting sports, football, running)
- Invasive procedures such as intubation, endoscopy, and/or colonoscopy unless essential because of reported tissue fragility of the trachea, esophagus, and small and large bowels
- Acetylsalicylate (aspirin) and long-term use of nonsteroidal anti-inflammatory drugs because of elevated risks of diverticulitis and diverticular bleeding
- The use of opioid medication for chronic pain, which does not lead to long-term pain relief and has the potential for addiction issues
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk relatives of an affected individual in order to identify as early as possible those who would benefit from prompt initiation of treatment and preventive measures.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
It is important that the obstetrician and midwives be made aware of the diagnosis of TNXB-related clEDS during a pregnancy. Reported pregnancy and postpartum issues in affected women include miscarriage, premature rupture of membranes, post- or peripartum hemorrhage, and prolapse of the rectum, vagina, and/or uterus [Green et al 2020].
Gynecologic follow up during pregnancy can be considered. Specialist delivery is strongly advised in view of the reported trachea rupture during intubation and esophagus rupture after insertion of a transesophageal ultrasound probe. These complications emphasize the need for careful handling of pregnant women with TNXB-related clEDS, especially in emergency situations [Brady et al 2017].
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
TNXB-related classical-like Ehlers-Danlos syndrome (clEDS) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected individual are obligate heterozygotes (i.e., presumed to be carriers of one TNXB pathogenic variant based on family history).
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for a TNXB pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent, the following possibilities should be considered:
- One of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017].
- Uniparental isodisomy for the parental chromosome with the pathogenic variant resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes may or may not be asymptomatic. Zweers et al [2003] investigated 20 heterozygous family members. All had significantly reduced serum TNX levels (compatible with heterozygote status). Clinical examination revealed generalized joint hypermobility and recurring joint dislocations and chronic joint pain in the majority of heterozygous females (nine of 14). Further investigations are needed to elucidate a possible role of reduced TNX levels and joint hypermobility. Skin hyperextensibility and easy bruising were absent in heterozygotes; heterozygotes as such do not fulfill the major criteria for TNXB-related clEDS and are not at risk of developing TNXB-related clEDS.
Sibs of a proband
- If both parents are known to be heterozygous for a TNXB pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being heterozygous, and a 25% chance of inheriting neither of the familial pathogenic variants.
- Although sibs with biallelic TNXB pathogenic variants are expected to fulfill the major criteria for TNXB-related clEDS, intrafamilial variability has been observed and the severity of related manifestations (e.g., musculoskeletal problems) can be variable.
- Heterozygotes may or may not be asymptomatic (see Parents of a proband).
Offspring of a proband. The offspring of an individual with TNXB-related clEDS are obligate heterozygotes (carriers) for a pathogenic variant in TNXB.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a TNXB pathogenic variant.
Carrier (Heterozygote) Detection
Carrier testing for at-risk relatives requires prior identification of the TNXB pathogenic variants in the family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Once the TNXB pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Ehlers-Danlos Society - EuropeUnited KingdomPhone: +44 203 887 6132
- Ehlers-Danlos Support UKUnited KingdomPhone: 0208 736 5604; 0800 9078518
- The Ehlers-Danlos SocietyPhone: 410-670-7577
- MedlinePlus
- DICE EDS and HSD Global Registry
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
TNXB-Related Classical-Like Ehlers-Danlos Syndrome: Genes and Databases
Table B.
OMIM Entries for TNXB-Related Classical-Like Ehlers-Danlos Syndrome (View All in OMIM)
Molecular Pathogenesis
TNXB encodes tenascin-X (TN-X), a glycoprotein of the extracellular matrix. Biallelic pathogenic variants result in a loss of the functional protein product and the characteristic clinical features. Pathogenic splice site or nonsense TNXB variants can lead to nonsense-mediated decay of the mutated RNA and result in complete absence of TN-X. Pathogenic missense variants may result in misfolding of the protein. Individuals with a molecularly confirmed diagnosis of TNXB-related classical-like Ehler-Danlos syndrome (clEDS) can have (1) intragenic pathogenic variants, (2) TNXB whole-gene or small deletions, or (3) contiguous deletions of TNXB and CYP21A2.
TNXB and CYP21A2 (associated with congenital adrenal hyperplasia) are located within the human leukocyte antigen histocompatibility complex at 6p23.1 alongside their homologous pseudogenes, TNXA and CYP21A1P. This region is prone to meiotic nonhomologous recombination resulting in gene conversions (exchange of DNA sequences between gene and pseudogene). Three types of TNXA/TNXB gene conversions have been identified [Morissette et al 2015]: (1) CAH-X chimera 1 (CH-1) has TNXB exons 35-44 replaced with TNXA and is characterized by the c.11435_11524+30del variant; (2) CAH-X chimera 2 (CH-2) has TNXB exons 40-44 replaced with TNXA and is characterized by the TNXA-derived c.12174C>G (p.Cys4058Trp) variant; (3) CAH-X chimera 3 (CH-3) has TNXB exons 41-44 replaced by TNXA and is characterized by three TNXA-derived missense variants. The exact pathogenic mechanism of the c.12174C>G (p.Cys4058Trp) variant is unknown, but the variant affects a cysteine residue that is predicted to form a disulfide bond, stabilizing tertiary protein structure [Demirdas et al 2017]. TNXB-related clEDS is often caused by gene conversion events, which are the most common recurrent mechanism of pathogenicity [Green et al 2020].
Mechanism of disease causation. Loss of function
TNXB-specific laboratory technical considerations. Molecular genetic testing may be complicated by the presence of pseudogene TNXA, which has greater than 97% homology to the 3' end (exons 32-44) of TNXB. As such, exon and intron sequences are (almost) identical in the gene and pseudogene [Demirdas et al 2017].
Table 8.
Notable TNXB Pathogenic Variants
Chapter Notes
Revision History
- 15 September 2022 (ma) Review posted live
- 24 September 2021 (fvd) Original submission
References
Literature Cited
- Besselink-Lobanova A, Maandag NJ, Voermans NC, van der Heijden HF, van der Hoeven JG, Heunks LM. Trachea rupture in tenascin-X-deficient type Ehlers-Danlos syndrome. Anesthesiology. 2010;113:746–9. [PubMed: 20693885]
- Brady AF, Demirdas S, Fournel-Gigleux S, Ghali N, Giunta C, Kapferer-Seebacher I, Kosho T, Mendoza-Londono R, Pope MF, Rohrbach M, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Zschocke J, Malfait F. The Ehlers-Danlos syndromes, rare types. Am J Med Genet C Semin Med Genet. 2017;175:70–115. [PubMed: 28306225]
- Brisset M, Metay C, Carlier RY, Badosa C, Marques C, Schalkwijk J, vanVlijmen-Willems I, Jimenez-Mallebrera C, Keren B, Jobic V, Laforêt P, Malfatti E. Biallelic mutations in Tenascin-X cause classical-like Ehlers-Danlos syndrome with slowly progressive muscular weakness. Neuromuscul Disord. 2020;30:833–8. [PubMed: 32988710]
- Burch GH, Gong Y, Liu W, Dettman RW, Curry CJ, Smith L, Miller WL, Bristow J. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat Genet. 1997;17:104–8. [PubMed: 9288108]
- Castori M, Voermans NC. Neurological manifestations of Ehlers-Danlos syndrome(s): a review. Iran J Neurol. 2014;13:190–208. [PMC free article: PMC4300794] [PubMed: 25632331]
- Chen W, Perritt AF, Morissette R, Dreiling JL, Bohn MF, Mallappa A, Xu Z, Quezado M, Merke DP. Ehlers Danlos syndrome caused by biallelic TNXB variants in patients with congenital adrenal hyperplasia. Hum Mutat. 2016;37:893–7. [PMC free article: PMC4983206] [PubMed: 27297501]
- Demirdas S, Dulfer E, Robert L, Kempers M, van Beek D, Micha D, van Engelen BG, Hamel B, Schalkwijk J, Loeys B, Maugeri A, Voermans NC. Recognizing the tenascin-X deficient type of Ehlers-Danlos syndrome: a cross-sectional study in 17 patients. Clin Genet. 2017;91:411–25. [PubMed: 27582382]
- Gbadegesin RA, Brophy PD, Adeyemo A, Hall G, Gupta IR, Hains D, Bartkowiak B, Rabinovich CE, Chandrasekharappa S, Homstad A, Westreich K, Wu G, Liu Y, Holanda D, Clarke J, Lavin P, Selim A, Miller S, Wiener JS, Ross SS, Foreman J, Rotimi C, Winn MP. TNXB mutations can cause vesicoureteral reflux. J Am Soc Nephrol. 2013;24:1313–22. [PMC free article: PMC3736717] [PubMed: 23620400]
- Ghali N, Baker D, Brady AF, Burrows N, Cervi E, Cilliers D, Frank M, Germain DP, Hulmes DJS, Jacquemont ML, Kannu P, Lefroy H, Legrand A, Pope FM, Robertson L, Vandersteen A, von Klemperer K, Warburton R, Whiteford M, van Dijk FS. Atypical COL3A1 variants (glutamic acid to lysine) cause vascular Ehlers-Danlos syndrome with a consistent phenotype of tissue fragility and skin hyperextensibility. Genet Med. 2019;21:2081–91. [PubMed: 30837697]
- Green C, Ghali N, Akilapa R, Angwin C, Baker D, Bartlett M, Bowen J, Brady AF, Brock J, Chamberlain E, Cheema H, McConnell V, Crookes R, Kazkaz H, Johnson D, Pope FM, Vandersteen A, Sobey G, van Dijk FS. Classical-like Ehlers-Danlos syndrome: a clinical description of 20 newly identified individuals with evidence of tissue fragility. Genet Med. 2020;22:1576–82. [PubMed: 32572181]
- Hendriks AGM, Voermans NC, Schalkwijk J, Hamel BC, van Rossum MM. Well-defined clinical presentation of Ehlers-Danlos syndrome in patients with tenascin-X deficiency: a report of four cases. Clin Dysmorphol. 2012;21:15–8. [PubMed: 21959861]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Lindor NM, Bristow J. Tenascin-X deficiency in autosomal recessive Ehlers-Danlos syndrome. Am J Med Genet A. 2005;135:75–80. [PubMed: 15793839]
- Malfait F, Castori M, Francomano CA, Giunta C, Kosho T, Byers PH. The Ehlers-Danlos syndromes. Nat Rev Dis Primers. 2020;6:64. [PubMed: 32732924]
- Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, Castori M, Cohen H, Colombi M, Demirdas S, De Backer J, De Paepe A, Fournel-Gigleux S, Frank M, Ghali N, Giunta C, Grahame R, Hakim A, Jeunemaitre X, Johnson D, Juul-Kristensen B, Kapferer-Seebacher I, Kazkaz H, Kosho T, Lavallee ME, Levy H, Mendoza-Londono R, Pepin M, Pope FM, Reinstein E, Robert L, Rohrbach M, Sanders L, Sobey GJ, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Wheeldon N, Zschocke J, Tinkle B. The 2017 international classification of the Ehlers–Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175:8–26. [PubMed: 28306229]
- Micale L, Guarnieri V, Augello B, Palumbo O, Agolini E, Sofia VM, Mazza T, Novelli A, Carella M, Castori M. Novel TNXB variants in two Italian patients with classical-like Ehlers-Danlos syndrome. Genes (Basel). 2019;10:967. [PMC free article: PMC6947605] [PubMed: 31775249]
- Miller WL, Merke DP. Tenascin-X, congenital adrenal hyperplasia, and the CAH-X syndrome. Horm Res Paediatr. 2018;89:352–61. [PMC free article: PMC6057477] [PubMed: 29734195]
- Morissette R, Chen W, Perritt AF, Dreiling JL, Arai AE, Sachdev V, Hannoush H, Mallappa A, Xu Z, McDonnell NB, Quezado M, Merke DP. Broadening the spectrum of Ehlers Danlos syndrome in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2015;100:E1143–52. [PMC free article: PMC4525000] [PubMed: 26075496]
- O'Connell M, Burrows NP, van Vlijmen-Willems MJJ, Clark SM. Schalkwijk. Tenascin-X deficiency and Ehlers-Danlos syndrome: a case report and review of the literature. Br J Dermatol. 2010;163:1340–5. [PubMed: 20649799]
- Pénisson-Besnier I, Allamand V, Beurrier P, Martin L, Schalkwijk J, van Vlijmen-Willems I, Gartioux C, Malfait F, Syx D, Macchi L, Marcorelles P, Arbeille B, Croué A, De Paepe A, Dubas F. Compound heterozygous mutations of the TNXB gene cause primary myopathy. Neuromuscul Disord. 2013;23:664–9. [PubMed: 23768946]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rymen D, Ritelli M, Zoppi N, Cinquina V, Giunta C, Rohrbach M, Colombi M. Clinical and molecular characterization of classical-like Ehlers-Danlos syndrome due to a novel TNXB variant. Genes (Basel). 2019;10:843. [PMC free article: PMC6895888] [PubMed: 31731524]
- Sakiyama T, Kubo A, Sasaki T, Yamada T, Yabe N, Matsumoto K, Futei Y. Recurrent gastrointestinal perforation in a patient with Ehlers-Danlos syndrome due to tenascin-X deficiency. J Dermatol. 2015;42:511–4. [PubMed: 25772043]
- Schalkwijk J, Zweers MC, Steijlen PM, Dean WB, Taylor G, van Vlijmen IM, van Haren B, Miller WL, Bristow J. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med. 2001;345:1167–75. [PubMed: 11642233]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Voermans NC, Drost G, van Kampen A, Gabreëls-Festen AA, Lammens M, Hamel BC, Schalkwijk J, van Engelen BG. Recurrent neuropathy associated with Ehlers-Danlos syndrome. J Neurol. 2006;253:670–1. [PubMed: 16311893]
- Voermans NC, Jenniskens GJ, Hamel BC, Schalkwijk J, Guicheney P, van Engelen BG. Ehlers-Danlos syndrome due to tenascin-X deficiency: muscle weakness and contractures support overlap with collagen VI myopathies. Am J Med Genet A. 2007;143A:2215–9. [PubMed: 17702048]
- Voermans NC, van Alfen N, Pillen S, Lammens M, Schalkwijk J, Zwarts MJ, van Rooij IA, Hamel BC, van Engelen BG. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol. 2009;65:687–97. [PubMed: 19557868]
- Watanabe S, Ito Y, Samura O, Nakano H, Sawamura D, Asahina A, Itoh M. Novel gross deletion mutation c.-105_4042+498del in the TNXB gene in a Japanese woman with classical-like Ehlers-Danlos syndrome: a case of uneventful pregnancy and delivery. J Dermatol. 2021;48:e227–e228. [PubMed: 33721335]
- Zweers MC, Bristow J, Steijlen PM, Dean WB, Hamel BC, Otero M, Kucharekova M, Boezeman JB, Schalkwijk J. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73:214–7. [PMC free article: PMC1180584] [PubMed: 12865992]
Publication Details
Author Information and Affiliations
London North West University Healthcare NHS Trust;
Department of Metabolism, Digestion and Reproduction
Genetics and Genomics Section
Imperial College London
London, United Kingdom
London North West University Healthcare NHS Trust;
Department of Metabolism, Digestion and Reproduction
Genetics and Genomics Section
Imperial College London
London, United Kingdom
Erasmus Medical Center
Erasmus University
Rotterdam, the Netherlands
Sheffield Children's NHS Foundation Trust
Sheffield, United Kingdom
Publication History
Initial Posting: September 15, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
van Dijk FS, Ghali N, Demirdas S, et al. TNXB-Related Classical-Like Ehlers-Danlos Syndrome. 2022 Sep 15. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.