Tyrosinemia Type II
Synonyms: Oculocutaneous Tyrosinemia, Richner-Hanhart Syndrome, TAT Deficiency, Tyrosine Aminotransferase Deficiency
Zahra Bayzaei, PhD, Seyed Mohsen Dehghani, MD, MPH, and Bita Geramizadeh, MD, APCP.
Author Information and AffiliationsInitial Posting: October 24, 2024.
Estimated reading time: 21 minutes
Summary
Clinical characteristics.
Tyrosinemia type II is characterized by corneal dystrophy, painful palmoplantar hyperkeratosis, and variable intellectual disability. Individuals diagnosed and treated from early infancy may be asymptomatic or have only mild ocular and skin manifestations. Individuals with delayed diagnosis or lack of treatment present with ocular, skin, and variable cognitive manifestations.
Diagnosis/testing.
The diagnosis of tyrosinemia type II is established in a proband by identification of biallelic pathogenic variants in TAT on molecular genetic testing or – in limited instances – significantly reduced activity of the enzyme tyrosine aminotransferase in liver.
Management.
Targeted therapy: Lifelong restriction of dietary tyrosine and phenylalanine with low-protein diet combined with age-appropriate amino acid (phenylalanine-free and tyrosine-free), vitamin, and mineral supplements.
Supportive care: Lubricating eye drops and ointment; ocular surgery as needed to treat bilateral corneal ulcers; pyridoxine phosphate or a systemic retinoid may be beneficial for skin manifestations; developmental and educational support.
Surveillance: Quantitative analysis of plasma tyrosine and phenylalanine concentrations at each visit; ophthalmology evaluation annually or as needed; evaluation by ophthalmic subspecialist annually or as recommended by ophthalmologist; skin assessment for hyperkeratotic plaques as clinically indicated; monitoring of developmental milestones, neuropsychological testing using age-appropriate standardized assessment batteries, and standardized quality-of-life assessment tools for affected individuals, parents, and/or caregivers annually or at each visit; measurement of plasma calcium, phosphorus, and 25-hydroxyvitamin D concentrations annually or as clinically indicated.
Agents/circumstances to avoid: Increased dietary protein.
Evaluation of relatives at risk: Testing of at-risk sibs of any age is warranted to allow for early diagnosis and treatment.
Genetic counseling.
Tyrosinemia type II is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a TAT pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the TAT pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives and prenatal/preimplantation genetic testing are possible.
Diagnosis
No consensus clinical diagnostic criteria for tyrosinemia type II have been published.
Suggestive Findings
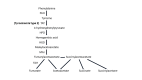
Tyrosinemia type II is caused by deficiency of the enzyme tyrosine aminotransferase (TAT) (see ). Tyrosinemia type II should be suspected in individuals with either abnormal newborn screening (NBS) results or clinical and laboratory findings suggestive of tyrosinemia type II. Note: Tyrosinemia type II is not typically included in NBS panels, but may be detected when screening for tyrosinemia type I.
The tyrosine catabolic pathway FAH = fumarylacetoacetate hydrolase; HGD = homogentisate 1,2-dioxygenase; HPD = 4-hydroxyphenylpyruvic acid dioxygenase; MAI = 4-maleylacetoacetate cis-trans-isomerase; PAH = phenylalanine hydroxylase; TAT = tyrosine aminotransferase (more...)
Scenario 1: Abnormal NBS Result
NBS for hepatorenal tyrosinemia (tyrosinemia type I) is typically based on quantification of tyrosine in dried blood spots. Elevated tyrosine above the cutoff reported by the screening laboratory is considered positive and requires follow-up biochemical testing. The identification of an infant with elevated tyrosine without the presence of succinylacetone (which is seen in tyrosinemia type I) should prompt investigation of other defects in tyrosine metabolism, including tyrosinemia type II.
Additional supportive laboratory findings of tyrosinemia type II include the following:
Markedly elevated plasma tyrosine concentration
Note: Elevated plasma tyrosine concentration can be a nonspecific indicator of liver damage or immaturity.
Normal succinylacetone measured directly from the newborn blood spot by tandem mass spectroscopy
Absence of succinylacetone in the blood and urine
Elevated urinary concentration of tyrosine metabolites (e.g., 4-hydroxyphenylpyruvate, 4-hydroxyphenyllactate, 4-hydroxyphenylacetate, N-acetyltyrosine, and 4-tyramine)
If the follow-up biochemical testing supports the likelihood of tyrosinemia type II, additional testing is required to confirm the diagnosis (see Establishing the Diagnosis).
Upon identification of high tyrosine levels on NBS (typically >500 µmol/L and may exceed 1,000 µmol/L), consultation with a metabolic physician / biochemical geneticist and specialist metabolic dietitian should be obtained while additional testing is performed to determine whether the result is a true positive NBS and to definitively establish the diagnosis of tyrosinemia type II.
Scenario 2: Symptomatic Individual
A symptomatic individual who has either (1) atypical findings associated with later-onset tyrosinemia type II or (2) untreated infantile-onset tyrosinemia type II may present with the following supportive clinical findings, preliminary laboratory findings, and family history.
Clinical findings
Ocular manifestations such as increased tearing, photophobia, pain, redness, and bilateral keratitis
Skin manifestations such as progressive, painful, nonpruritic, and hyperkeratotic plaques on soles and palms, often associated with hyperhidrosis; usually begin after first year of life
Developmental delay, particularly in those with high blood tyrosine concentration
Variable intellectual disability
Laboratory findings
Elevated plasma tyrosine concentration
Elevated 4-hydroxyphenylpyruvate, 4-hydroxyphenyllactate, and 4-hydroxyphenylacetate and presence of small quantities of N-acetyltyrosine and 4-tyramine on urine organic acid analysis.
Serum transaminase levels are usually normal.
Note: Because these laboratory findings are not specific to tyrosinemia type II, additional testing is required to establish the diagnosis.
Family history may suggest autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not exclude the diagnosis.
Establishing the Diagnosis
The diagnosis of tyrosinemia type II is established in a proband by identification of biallelic pathogenic (or likely pathogenic) variants in TAT on molecular genetic testing (see Table 1) or – in limited instances – significantly reduced activity of the enzyme tyrosine aminotransferase in liver. Because of its relatively high sensitivity, TAT molecular genetic testing can obviate the need for enzymatic testing and, thus, is increasingly the preferred confirmatory test for tyrosinemia type II [Beyzaei et al 2022].
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of biallelic TAT variants of uncertain significance (or of one known TAT pathogenic variant and one TAT variant of uncertain significance) does not establish or rule out the diagnosis.
Scenario 1: Abnormal newborn screening (NBS) result. When NBS results and other laboratory findings suggest the diagnosis of tyrosinemia type II, molecular genetic testing approaches can include single-gene testing or use of a multigene panel.
Single-gene testing. Sequence analysis of TAT is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
A multigene panel that includes
TAT and other genes of interest (see
Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Scenario 2: A symptomatic individual. When the diagnosis of tyrosinemia type II has not been considered (because an individual has atypical findings associated with later-onset tyrosinemia type II or untreated infantile-onset tyrosinemia type II resulting from NBS not performed or false negative NBS result), comprehensive
genomic
testing (which does not require the clinician to determine which gene[s] are likely involved) is an option. Exome sequencing is most commonly used; genome sequencing is also possible. To date, the majority of TAT pathogenic variants reported (e.g., missense, nonsense) are within the coding region and are likely to be identified on exome sequencing.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Tyrosinemia Type II
View in own window
| Gene 1 | Method | Proportion of Pathogenic Variants 2 Identified by Method |
|---|
|
TAT
| Sequence analysis 3 | >95% 4 |
| Gene-targeted deletion/duplication analysis 5 | <5% 4, 6 |
- 1.
- 2.
- 3.
Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Variants may include missense, nonsense, and splice site variants and small intragenic deletions/insertions; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
- 4.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications. Exome and genome sequencing may be able to detect deletions/duplications using breakpoint detection or read depth; however, sensitivity can be lower than gene-targeted deletion/duplication analysis.
- 6.
Clinical Characteristics
Clinical Description
Tyrosinemia type II is characterized by corneal dystrophy, painful palmoplantar hyperkeratosis, and variable intellectual disability. Individuals diagnosed and treated from early infancy may be asymptomatic or have only mild ocular and skin manifestations. Individuals with delayed diagnosis or lack of treatment present with ocular, skin, and variable cognitive manifestations [Gliagias et al 2022]. To date, more than 70 individuals have been identified with tyrosinemia type II [Beyzaei et al 2022]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Select Features of Tyrosinemia Type II
View in own window
| Feature | % of Persons w/Feature | Comment |
|---|
| Diagnosis & treatment in early infancy | Later diagnosis w/delayed or no treatment |
|---|
|
Ocular manifestations
| 75% | ~75%-85% | ↑ tearing, photophobia, eye redness, dendritic epithelial ulcers |
|
Skin manifestations
| 35% | 80% | Palmoplantar hyperkeratosis, hyperkeratotic plaques |
|
Intellectual disability
| Rare | ~60% | |
|
Developmental delay
| Rare | ~10% | |
Ocular manifestations can appear in the first months or first year of life or as late as the fourth decade [Viglizzo et al 2006]. Ocular symptoms include epiphora (increased tearing), photophobia, pain, and blepharospasm. Ocular signs include corneal clouding, pseudodendritic corneal lesions (usually bilateral), dendritic ulcers, and, rarely, corneal or conjunctival plaques. The dendritic lesions on the cornea stain poorly with fluorescein.
Bacterial, viral, and fungal cultures are typically negative.
Ocular manifestations may undergo spontaneous remission and/or recurrence and may occur independently of other clinical manifestations.
Individuals are often misdiagnosed with herpes simplex keratitis [
Soares et al 2017].
Bilateral corneal lesions unresponsive to antiviral therapies should alert the clinician to the possible diagnosis of tyrosinemia type II, even in the absence of skin lesions [
Macsai et al 2001].
Some individuals with tyrosinemia type II report isolated burning and redness of the eyes since childhood [
Ghalamkarpour et al 2020].
Asynchronous bilateral eye disease was the sole clinical feature reported in one child age 15 months with negative NBS for tyrosinemia type II [
Gliagias et al 2022]. Following diet modification, tyrosine levels decreased, resulting in significant resolution of ocular symptoms [
Gliagias et al 2022].
Corneal lesions recurred in one individual after discontinuation of dietary restrictions [
Macsai et al 2001].
Skin manifestations can appear in the first months of life or be delayed until the second decade [Rabinowitz et al 1995]. They typically present as progressive painful hyperkeratotic plaques on the soles and palms, often associated with hyperhidrosis [da Silva et al 2024]. The fingertips and weightbearing areas of the soles are commonly affected by keratoderma.
Developmental delay. Infants diagnosed and treated in early infancy typically have normal development. Untreated individuals can have global developmental delay including poor feeding with poor appetite and reduced oral intake [Gliagias et al 2022].
Mild developmental delay with hemiparesis and seizures has been reported in one individual [Charfeddine et al 2006].
Intellectual disability. About half of individuals with tyrosinemia type II have mild intellectual disability, but early dietary treatment may reduce this risk [Legarda et al 2011, Nakamura et al 2015, da Silva et al 2024]. Mild language disorder has been reported in three individuals [Charfeddine et al 2006]. Intellectual disability may be prevented by early dietary restriction of tyrosine and phenylalanine. Language development was reported to improve with dietary treatment [Tsai et al 2006].
Nutritional deficiencies can appear in individuals due to limitations of certain amino acids (phenylalanine, tyrosine, and methionine) in the diet. Deficiency of calcium, phosphorus, and 25-hydroxyvitamin D have been observed.
Other
Isolated geographic tongue with otherwise normal clinical findings (1 individual) [
Bouyacoub et al 2013]
Self-harm (agitation with paroxysms of head banging and hand and tongue biting) and diffuse plantar keratoderma (1 individual) [
Madan & Gupta 2006]
Genotype-Phenotype Correlations
Precise genotype-phenotype correlations are difficult to determine, as most TAT pathogenic variants reported to date are not recurrent [Beyzaei et al 2022].
The most common variant reported to date, p.Arg57Ter, has been reported in ten affected individuals from three families [Peña-Quintana et al 2017, Beyzaei et al 2022]. Individuals homozygous for this pathogenic variant typically present in the neonatal period and have eye and skin manifestations.
Nomenclature
Tyrosinemia type II was previously referred to as keratosis palmoplantaris with corneal dystrophy.
Prevalence
Tyrosinemia type II is rare, with an incidence of less than one in 250,000 [Bouyacoub et al 2013]. Fewer than 100 affected individuals from various ethnic backgrounds have been reported to date [Janakiraman et al 2006, El-Shabrawi & Kamal 2013].
Founder pathogenic variants have been reported in populations from northern Italy (Lombardy and Tuscany; p.Arg57Ter), Tunisia (p.Cys151Tyr), Lebanon (p.Arg297Ter), Palestine (p.Arg417Ter), and Gran Canaria (p.Pro406Leu), which has a population of Mediterranean ancestry primarily from continental Spain and northern Africa [Peña-Quintana et al 2017, Beyzaei et al 2022]. Consanguinity is frequently reported in parents of affected individuals, which may contribute to the higher prevalence of this disorder among Arab populations [Charfeddine et al 2006].
Differential Diagnosis
Hypertyrosinemia. Other causes of hypertyrosinemia include genetic tyrosine metabolism disorders (see Table 3) and acquired conditions such as the following:
Corneal lesions. Because ocular features are often the initial manifestations of tyrosinemia type II, pseudodendritic keratitis is often mistaken for herpes simplex keratitis.
Genetic disorders of interest in the differential diagnosis of tyrosinemia type II are listed in Table 3.
Table 3.
Genes of Interest in the Differential Diagnosis of Tyrosinemia Type II
View in own window
| Gene(s) | Disorder | MOI | Features of Disorder |
|---|
| Overlapping w/tyrosinemia type II | Distinguishing from tyrosinemia type II |
|---|
|
FAH
|
Tyrosinemia type I
| AR | Hypertyrosinemia | Liver & kidney dysfunction ↑ succinylacetone concentration in blood & urine Hypoglycemia High alkaline phosphatase Moderately ↑ plasma concentration of tyrosine, phenylalanine, & other amino acids, esp methionine (due to secondary inhibition of methionine adenosyltransferase)
|
|
HPD
| Tyrosinemia type III (OMIM 276710) | AR | Hypertyrosinemia | Moderately ↑ plasma concentration of tyrosine Low activity of enzyme HPD in liver biopsy Ataxia & seizures Absence of skin & ocular lesions
|
MBTPS2
PERP
TRPV3
| Olmsted syndrome (OMIM PS614594) | AD
XL | Palmoplantar keratoderma w/periorificial keratotic plaques |
|
AD = autosomal dominant; AR = autosomal recessive; HPD = 4-hydroxyphenylpyruvic acid dioxygenase; MOI = mode of inheritance; XL = X-linked
Management
No clinical practice guidelines for tyrosinemia type II have been published. In the absence of published guidelines, the following recommendations are based on the authors' personal experience managing individuals with this disorder.
When tyrosinemia type II is suspected during the diagnostic evaluation due to high levels of tyrosine (typically >500 µmol/L and may exceed 1,000 µmol/L), metabolic treatment should be initiated immediately (see Targeted Therapy).
Development and evaluation of treatment plans, training and education of affected individuals and their families, and avoidance of side effects of dietary treatment (i.e., malnutrition, growth failure) require a multidisciplinary approach including multiple subspecialists, with oversight and expertise from a specialized metabolic center and the involvement of a dietitian with specialized training.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with tyrosinemia type II, the evaluations summarized Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Tyrosinemia Type II: Recommended Evaluations Following Initial Diagnosis
View in own window
| System/Concern | Evaluation | Comment |
|---|
|
Eyes
| Ophthalmologic eval | To assess for epiphora, photophobia, & blepharospasm & more complex findings (e.g., corneal clouding, corneal lesions, & dendritic ulcers) that may require referral for subspecialty care &/or low vision services |
|
Skin
| Skin exam | To assess for hyperkeratotic plaques on soles, palms (esp thenar & hypothenar areas), & fingertips |
|
Development
| Developmental assessment | To incl motor, adaptive, cognitive, & speech-language eval Eval for early intervention / special education
|
|
Metabolic
| Consultation w/metabolic physician / biochemical geneticist & specialist metabolic dietitian | To initiate dietary restriction of tyrosine & phenylalanine |
|
Genetic counseling
| By genetics professionals 1 | To obtain a pedigree & inform affected persons & their families re nature, MOI, & implications of tyrosinemia type II to facilitate medical & personal decision making |
Family support
& resources
| By clinicians, wider care team, & family support organizations | Assessment of family & social structure to determine need for:
|
MOI = mode of inheritance
- 1.
Medical geneticist, certified genetic counselor, certified advanced genetic nurse
Treatment of Manifestations
Targeted Therapy
In GeneReviews, a targeted therapy is one that addresses the specific underlying mechanism of disease causation (regardless of whether the therapy is significantly efficacious for one or more manifestation of the genetic condition); would otherwise not be considered without knowledge of the underlying genetic cause of the condition; or could lead to a cure. —ED
Restriction of dietary tyrosine and phenylalanine. Targeted therapy for tyrosinemia type II aims to reduce plasma tyrosine concentration and consists of a low-protein diet with age-appropriate amino acid (tyrosine- and phenylalanine-free), vitamin, and mineral supplements.
Treatment is a lifelong tyrosine- and phenylalanine-restricted diet. Initially this is in the form of a prescribed infant formula. After weaning, a low-protein diet is prescribed and age-appropriate amino acid (tyrosine- and phenylalanine-free), vitamin, and mineral supplements are given.
A diet that is low in tyrosine and phenylalanine can help correct chemical abnormalities and lead to significant improvement in skin and eye lesions.
Supportive Care
Supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 5).
Table 5.
Tyrosinemia Type II: Treatment of Manifestations
View in own window
| Principle/Manifestation | Treatment | Considerations/Other |
|---|
|
Ocular manifestations
|
| Ocular manifestations improve & may resolve w/dietary restriction of tyrosine & phenylalanine (see Targeted Therapy). |
|
Skin manifestations
| Pyridoxine phosphate or systemic retinoid may be beneficial. | Skin manifestations often resolve w/dietary restriction of tyrosine & phenylalanine (see Targeted Therapy). |
Developmental delay /
Intellectual disability
| Developmental & educational support as needed | |
Surveillance
In addition to regular evaluations by a metabolic specialist and metabolic dietician, the evaluations summarized in Table 6 are recommended to monitor existing manifestations, the individual's response to targeted therapy and supportive care, and the emergence of new manifestations.
Table 6.
Tyrosinemia Type II: Recommended Surveillance
View in own window
| Manifestation | Evaluation | Frequency/Comment |
|---|
Metabolic/
Nutrition
| Quantitative analysis of plasma tyrosine & phenylalanine concentrations | At each visit |
|
Ocular manifestations
| Eval by ophthalmologist to assess for epiphora, photophobia, blepharospasm, & keratitis | Annually or as needed |
| Eval by ophthalmic subspecialist to assess for more complex findings (e.g., corneal clouding, pseudodendritic corneal lesions, dendritic ulcers, & corneal or conjunctival plaques) | Annually or as recommended by ophthalmologist |
|
Skin manifestations
| Skin assessment for hyperkeratotic plaques | As clinically indicated |
|
Delayed acquisition of developmental milestones
| Monitoring of developmental milestones Neuropsychological testing using age-appropriate standardized assessment batteries Standardized quality-of-life assessment tools for affected persons & parents/caregivers
| Annually or at each visit |
|
Nutritional deficiencies
| Measurement of plasma calcium, phosphorus, & 25-hydroxyvitamin D concentrations | Annually or as clinically indicated |
Agents/Circumstances to Avoid
Avoid increased dietary protein.
Evaluation of Relatives at Risk
Testing of at-risk sibs of any age is warranted to allow for early diagnosis and treatment of tyrosinemia type II.
Prenatal testing of a fetus at risk. When the pathogenic variants causing tyrosinemia type II in the family are known, prenatal testing of fetuses at risk may be performed via amniocentesis or chorionic villus sampling to facilitate institution of treatment at birth.
Newborn sib. If prenatal testing was not performed – in parallel with newborn screening – testing for the familial TAT pathogenic variants or measurement of plasma tyrosine concentration and excretion of 4-hydroxyphenylpyruvate, 4-hydroxyphenyllactate, and 4-hydroxyphenylacetate on urine organic acid analysis can be performed.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Infants born to women with untreated tyrosinemia type II may have an increased risk of intrauterine growth deficiency and developmental delay [Cerone et al 2002]. Careful dietary control of plasma tyrosine concentration during pregnancy is recommended.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Tyrosinemia type II is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
Sibs of a proband
If both parents are known to be heterozygous for a TAT pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. Unless an affected individual's reproductive partner also has tyrosinemia type II or is a carrier, offspring will be obligate heterozygotes (carriers) for a pathogenic variant in TAT.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a TAT pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the TAT pathogenic variants in the family.
Prenatal Testing and Preimplantation Genetic Testing
Once the TAT pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal and preimplantation genetic testing. While most health care professionals would consider use of prenatal and preimplantation genetic testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Tyrosinemia Type II: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
TAT encodes the enzyme tyrosine aminotransferase (TAT), the first in a series of five enzymes that catalyze reversible transamination of tyrosine to 4-hydroxyphenylpyruvate. Reduced TAT function results in the accumulation of upstream byproducts of tyrosine and phenylalanine.
TAT is a homodimer and is composed of two identical polypeptide chains. It is expressed in the liver, kidneys, and brain. The enzyme has the highest activity in the liver. In TAT, the first 38 amino acids may not be involved in enzyme dimerization and are not required in the active site stability and enzyme-substrate interactions. This N-terminal fragment, however, is required for targeting by the ubiquitin-proteasome pathway, which degrades proteins to small peptides.
Mechanism of disease causation. Loss of function
Table 7.
TAT Pathogenic Variants Referenced in This GeneReview
View in own window
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Chapter Notes
Author Notes
Dr Bita Geramizadeh is a molecular pathologist. Her clinical and research interests include transplant hepatology, acute liver failure, and inherited metabolic disorders of the liver. She is the author of numerous (more than 450) original manuscripts and reviews on the subject. Her research has been in both the clinical and basic science spheres. Dr Geramizadeh has launched and established a transplant research center and national registry trial for inherited metabolic disorders with a data coordinating center at the Shiraz University of Medical Sciences. She is the vice president of the Association for Inherited Metabolic Disorders (Ibn Sina).
Dr Seyed Mohsen Dehghani is a professor of pediatric gastroenterology and hepatology with experience in inherited disorders and more than 200 highly cited published papers.
Dr Zahra Beyzaei is a molecular and biochemical geneticist. She is a staff scientist in the transplant research center at the Shiraz University of Medical Sciences. She would be happy to communicate with persons who have any questions regarding tyrosinemia type II disease. Contact Dr Beyzaei to inquire about review of TAT variants of uncertain significance.
Acknowledgments
We extend our gratitude to the individuals with tyrosinemia type II and their families, our institutions, laboratory personnel, and researchers, as well as the Association for Inherited Metabolic Disorders (Ibn Sina), for their dedicated efforts and collaborative work.
References
Literature Cited
Beyzaei
Z, Nabavizadeh
S, Karimzadeh
S, Geramizadeh
B. The mutation spectrum and ethnic distribution of non-hepatorenal tyrosinemia (types II, III).
Orphanet J Rare Dis.
2022;17:424.
[
PMC free article: PMC9724276] [
PubMed: 36471409]
Bouyacoub
Y, Zribi
H, Azzouz
H, Nasrallah
F, Abdelaziz
RB, Kacem
M, Rekaya
B, Messaoud
O, Romdhane
L, Charfeddine
C, Bouziri
M, Bouziri
S, Tebib
N, Mokni
M, Kaabachi
N, Boubaker
S, Abdelhak
S. Novel and recurrent mutations in the TAT gene in Tunisian families affected with Richner-Hanhart syndrome.
Gene.
2013;529:45-9.
[
PubMed: 23954227]
Cerone
R, Fantasia
AR, Castellano
E, Moresco
L, Schiaffino
MC, R. G. Pregnancy and tyrosinaemia type II.
J Inherit Metab Dis.
2002;25:317-18.
[
PubMed: 12227462]
Charfeddine
C, Monastiri
K, Mokni
M, Laadjimi
A, Kaabachi
N, Perin
O, Nilges
M, Kassar
S, Keirallah
M, Guediche
MN, Kamoun
MR, Tebib
N, Ben Dridi
MF, Boubaker
S, Ben Osman
A, Abdelhak
S. Clinical and mutational investigations of tyrosinemia type II in northern Tunisia: identification and structural characterization of two novel TAT mutations.
Mol Genet Metab.
2006;88:184-91.
[
PubMed: 16574453]
da Silva
ISF, Sopa
I, Gomes
D, Peixoto
L, Oliveira
A. From skin lesions to tyrosinemia type II diagnosis.
J Inherit Metab Dis.
2024. Epub ahead of print.
[
PubMed: 38408363]
El-Shabrawi
M, Kamal
NM. Current management options for tyrosinemia. Orphan Drugs: Research and Reviews.
2013;3:1-9.
Ghalamkarpour
F, Niknezhad
N, Niknejad
N.
Familial Richner-Hanhart syndrome: report of a sibling with incomplete presentation.
Dermatol Ther.
2020;33:e14072.
[
PubMed: 32713067]
Janakiraman
L, Sathiyasekaran
M, Deenadayalan
M, Ganesh
R, Mahesh
U.
Richner Hanhart syndrome.
Indian J Pediatr.
2006;73:161-2.
[
PubMed: 16514229]
Jónsson
H, Sulem
P, Kehr
B, Kristmundsdottir
S, Zink
F, Hjartarson
E, Hardarson
MT, Hjorleifsson
KE, Eggertsson
HP, Gudjonsson
SA, Ward
LD, Arnadottir
GA, Helgason
EA, Helgason
H, Gylfason
A, Jonasdottir
A, Jonasdottir
A, Rafnar
T, Frigge
M, Stacey
SN, Th Magnusson
O, Thorsteinsdottir
U, Masson
G, Kong
A, Halldorsson
BV, Helgason
A, Gudbjartsson
DF, Stefansson
K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland.
Nature.
2017;549:519-22.
[
PubMed: 28959963]
Legarda
M, Wlodarczyk
K, Lage
S, Andrade
F, Kim
GJ, Bausch
E, Scherer
G, LJ. A-E. A large TAT deletion in a tyrosinaemia type II patient.
Mol Genet Metab.
2011;104:407-9.
[
PubMed: 21636300]
Macsai
MS, Schwartz
TL, Hinkle
D, Hummel
MB, Mulhern
MG, Rootman
D. Tyrosinemia type II: nine cases of ocular signs and symptoms.
Am J Ophthalmol.
2001;132:522-7.
[
PubMed: 11589874]
Madan
V, Gupta
U.
Tyrosinaemia type II with diffuse plantar keratoderma and self-mutilation.
Clin Exp Dermatol.
2006;31:54-6.
[
PubMed: 16309482]
Maydan
G, Andresen
BS, Madsen
PP, Zeigler
M, Raas-Rothschild
A, Zlotogorski
A, Gutman
A, Korman
SH. TAT gene mutation analysis in three Palestinian kindreds with oculocutaneous tyrosinaemia type II; characterization of a silent exonic transversion that causes complete missplicing by exon 11 skipping.
J Inherit Metab Dis.
2006;29:620-6.
[
PubMed: 16917729]
Murphy E. Tyrosinaemia type I. In: CEM Hollak, R Lachmann, eds. Inherited Metabolic Disease in Adults: A Clinical Guide. New York: Oxford Academic;2016:chap. 14.
Nakamura
K, Matsumoto
S, Mitsubuchi
H, Endo
F. Diagnosis and treatment of hereditary tyrosinemia in Japan.
Pediatr Int.
2015;57:37-40.
[
PubMed: 25443793]
Peña-Quintana
L, Scherer
G, Curbelo-Estévez
ML, Jiménez-Acosta
F, Hartmann
B, La Roche
F, Meavilla-Olivas
S, Pérez-Cerdá
C, García-Segarra
N, Giguère
Y, Huppke
P, Mitchell
GA, Mönch
E, Trump
D, Vianey-Saban
C, Trimble
ER, Vitoria-Miñana
I, Reyes-Suárez
D, Ramírez-Lorenzo
T, Tugores
A. Tyrosinemia type II: mutation update, 11 novel mutations and description of 5 independent subjects with a novel founder mutation.
Clin Genet.
2017;92:306-17.
[
PubMed: 28255985]
Rabinowitz
LG, Williams
LR, Anderson
CE, Mazur
A, Kaplan
P. Painful keratoderma and photophobia: hallmarks of tyrosinemia type II.
J Pediatr.
1995;126:266-9.
[
PubMed: 7844676]
Richards
S, Aziz
N, Bale
S, Bick
D, Das
S, Gastier-Foster
J, Grody
WW, Hegde
M, Lyon
E, Spector
E, Voelkerding
K, Rehm
HL, et al.
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Genet Med.
2015;17:405-24.
[
PMC free article: PMC4544753] [
PubMed: 25741868]
Soares
DC, Stroparo
MN, Lian
YC, Takakura
CY, Wolf
S, Betz
R, Kim
CA. Herpetiform keratitis and palmoplantar hyperkeratosis: warning signs for Richner-Hanhart syndrome.
J Inherit Metab Dis.
2017;40:461-2.
[
PubMed: 27832414]
Stenson
PD, Mort
M, Ball
EV, Chapman
M, Evans
K, Azevedo
L, Hayden
M, Heywood
S, Millar
DS, Phillips
AD, Cooper
DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting.
Hum Genet.
2020;139:1197-207.
[
PMC free article: PMC7497289] [
PubMed: 32596782]
Tekin
B, Yucelten
D, Zeybek
CA, Kiykim
E, Wehner
M, Betz
RC, Toker
AE. Oculocutaneous tyrosinemia: a case report with delayed diagnosis and excellent response to dietary modification.
Indian J Dermatol Venereol Leprol.
2015;81:303-5.
[
PubMed: 25784227]
Tsai
CP, Lin
PY, Lee
NC, Niu
DM, Lee
SM, Hsu
WM. Corneal lesion as the initial manifestation of tyrosinemia type II.
J Chin Med Assoc.
2006;69:286-8.
[
PubMed: 16863017]
Viglizzo
GM, Occella
C, Bleidl
D, Rongioletti
F. Richner-Hanhart syndrome (tyrosinemia II): early diagnosis of an incomplete presentation with unusual findings.
Pediatr Dermatol.
2006;23:259-61.
[
PubMed: 16780475]