Summary
Clinical characteristics.
Untreated tyrosinemia type I usually presents either in young infants with severe liver involvement or later in the first year with liver dysfunction and renal tubular dysfunction associated with growth failure and rickets. Untreated children may have repeated, often unrecognized, neurologic crises lasting one to seven days that can include change in mental status, abdominal pain, peripheral neuropathy, and/or respiratory failure requiring mechanical ventilation. Death in the untreated child usually occurs before age ten years, typically from liver failure, neurologic crisis, or hepatocellular carcinoma. Combined treatment with nitisinone and a low-tyrosine diet has resulted in a greater than 90% survival rate, normal growth, improved liver function, prevention of cirrhosis, correction of renal tubular acidosis, and improvement in secondary rickets.
Diagnosis/testing.
Tyrosinemia type I results from deficiency of the enzyme fumarylacetoacetase (FAH). The diagnosis is established in a proband with typical biochemical findings (increased succinylacetone concentration in the blood and urine; elevated plasma concentrations of tyrosine, methionine, and phenylalanine; and elevated urinary concentration of tyrosine metabolites and the compound δ-ALA) and/or by the identification of biallelic pathogenic variants in FAH on molecular genetic testing.
Management.
Treatment of manifestations: Nitisinone (Orfadin®), 2-(2-nitro-4-trifluoro-methylbenzyol)-1,3 cyclohexanedione (NTBC), which blocks parahydroxyphenylpyruvic acid dioxygenase (p-HPPD), the second step in the tyrosine degradation pathway, prevents the accumulation of fumarylacetoacetate and its conversion to succinylacetone. Nitisinone treatment should begin as soon as the diagnosis of tyrosinemia type I is confirmed. Because nitisinone increases the blood concentration of tyrosine, dietary management with controlled intake of phenylalanine and tyrosine should be started immediately after diagnosis to prevent tyrosine crystals from forming in the cornea. If the blood concentration of phenylalanine becomes too low (<20 μmol/L), additional natural protein should be added to the diet. Prior to the availability of nitisinone, the only definitive therapy for tyrosinemia type I was liver transplantation, which now should be reserved for those children who have severe liver failure at presentation and fail to respond to nitisinone therapy or have documented evidence of malignant changes in hepatic tissue.
Prevention of primary manifestations: Initiation of treatment with nitisinone as soon as the diagnosis is confirmed.
Prevention of secondary complications: Treatment of early signs of carnitine deficiency, osteoporosis, and rickets that are secondary to renal tubular Fanconi syndrome.
Surveillance: Guidelines for routine surveillance of individuals with tyrosinemia type I have been established.
Agents/circumstances to avoid: Inappropriate protein intake.
Evaluation of relatives at risk: All subsequent children of the parents of a child with tyrosinemia type I should have urine and blood succinylacetone analyzed as soon as possible after birth to enable the earliest possible diagnosis and initiation of therapy. If the pathogenic variants in the family are known, prenatal molecular genetic testing of an at-risk pregnancy may be considered.
Pregnancy management: Little data exist on the use of nitisinone during human pregnancy; however, at least two women have given birth to healthy infants while receiving therapeutic doses of nitisinone.
Genetic counseling.
Tyrosinemia type I is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk relatives and prenatal testing for a pregnancy at increased risk are possible if both pathogenic variants in a family are known.
Diagnosis
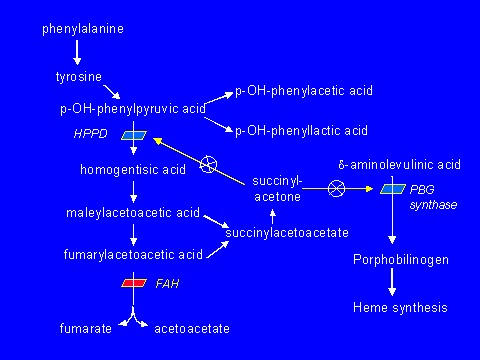
Tyrosinemia type I is caused by deficiency of the enzyme fumarylacetoacetase (FAH) (EC 3.7.1.2) (see Figure 1 and Pathophysiology).

Figure 1.
The tyrosine catabolic pathway
Suggestive Findings
Tyrosinemia type I should be suspected in individuals with the following newborn screening results, clinical features, and supportive laboratory findings.
Newborn screening
- Presence of succinylacetone, measured directly from the newborn blood spot by tandem mass spectroscopy, is pathognomonic for tyrosinemia type 1.
- Elevated tyrosine or methionine concentration in the blood suggests liver disease, which can be from a variety of causes; the diagnosis of tyrosinemia type I should be further evaluated by quantification of plasma or urinary succinylacetone.
- Infants with tyrosinemia type I may have only modestly elevated or normal blood concentrations of tyrosine and methionine when the first newborn screening sample is collected.
- Elevated tyrosine concentration on newborn screening can be the result of transient tyrosinemia of the newborn, tyrosinemia type II or III, or other liver disease.
- Elevated methionine concentration can indicate liver dysfunction, defects in methionine metabolism, or homocystinuria (see Homocystinuria Caused by Cystathionine Beta-Synthetase Deficiency).
- Low delta-ALA-dehydratase (PBG synthase) enzyme activity. This is measured in the newborn screening program in Quebec, Canada [Giguère et al 2005]. Succinylacetone is then measured in the urine of infants with apparent δ-ALA dehydratase deficiency [Schulze et al 2001].
Clinical features (in untreated individuals)
- Severe liver disease in young infants
- Signs of renal disease, rickets, and/or neurologic crises in children older than age six monthsUntreated children may have repeated neurologic crises lasting one to seven days that can include change in mental status, abdominal pain, peripheral neuropathy, and/or respiratory failure requiring mechanical ventilation.
Supportive laboratory findings
- Increased succinylacetone concentration in the blood and excretion in the urineNote: (1) Increased excretion of succinylacetone in the urine of a child with liver failure or severe renal disease is a pathognomonic sign of tyrosinemia type I. (2) Many laboratories require that measurement of succinylacetone be specifically requested when ordering urine organic acids.
- Elevated plasma concentration of tyrosine, methionine, and phenylalanineNote: (1) Plasma tyrosine concentration in affected infants can be normal in cord blood and during the newborn period. (2) Elevated plasma tyrosine concentration can also be a nonspecific indicator of liver damage or immaturity; for example, in infants taking a high-protein formula [Techakittiroj et al 2005], including undiluted goat's milk [Hendriksz & Walter 2004].
- Elevated urinary concentration of tyrosine metabolites p-hydroxyphenylpyruvate, p-hydroxyphenyllactate, and p-hydroxyphenylacetate detected on urine organic acid testing
- Increased urinary excretion of the compound δ-ALA secondary to inhibition of the enzyme δ-ALA dehydratase by succinylacetone in the liver and circulating red blood cells [Sassa & Kappas 1983]
- Changes in liver function (in untreated tyrosinemia type 1)
- Markedly elevated serum concentration of alpha-fetoprotein (average 160,000 ng/mL) (normal: <1,000 ng/mL for infants age 1-3 months; <12 ng/mL for children age 3 months to 18 years)
- Prolonged prothrombin and partial thromboplastin times
Note: (1) Changes in serum concentration of alpha-fetoprotein (AFP) and prothrombin time / partial thromboplastin time (PT/PTT) are more severe in tyrosinemia type I than in nonspecific liver disease and are often the presenting findings in tyrosinemia type I. (2) Transaminases and bilirubin are only modestly elevated, if at all. (3) An individual with liver disease and normal serum concentration of AFP and normal PT/PTT has a low probability of having tyrosinemia type I.
Establishing the Diagnosis
The diagnosis of tyrosinemia type I is established in a proband with characteristic biochemical findings (increased succinylacetone concentration in the blood and urine; elevated plasma concentrations of tyrosine, methionine, and phenylalanine; and elevated urinary concentration of tyrosine metabolites and the compound δ-ALA) and/or by identification of biallelic pathogenic variants in FAH on molecular genetic testing (see Table 1).
Molecular Genetic Testing
Single-gene testing. Sequence analysis of FAH is performed first and followed by gene-targeted deletion/duplication analysis if only one or no pathogenic variant is found.
- Targeted analysis for the p.Pro261Leu pathogenic variant can be performed first in individuals of Ashkenzai Jewish ancestry; this variant accounts for more than 99% of the pathogenic variants in this population [Elpeleg et al 2002].
- The pathogenic variant c.1062+5G>A (IVS12+5 G>A) accounts for 87.9% of pathogenic variants in the French Canadian population [Poudrier et al 1996].
- The four common FAH pathogenic variants – c.1062+5G>A (IVS12+5 G>A), c.554-1G>T (IVS6-1 G>T), c.607-6T>G (IVS7-6 T>G), and p.Pro261Leu – account for approximately 60% of pathogenic variants in tyrosinemia type I in the general US population [CR Scott, unpublished data].
Table 1.
Molecular Genetic Testing Used in Tyrosinemia Type I
Clinical Characteristics
Clinical Description
For children who were not detected by newborn screening, tyrosinemia type I usually presents either in young infants with severe liver involvement or later in the first year with liver dysfunction and significant renal involvement, growth failure, and rickets. Growth failure results from chronic illness with poor nutritional intake, liver involvement, and/or chronic renal disease. Death in the undetected or untreated child usually occurs before age ten years, typically from liver failure, neurologic crisis, or hepatocellular carcinoma.
- Liver involvement. Undetected or untreated children presenting before age six months typically have acute liver failure with initial loss of synthetic function for clotting factors. PT and PTT are markedly prolonged and not corrected by vitamin K supplementation; factor II, VII, IX, XI, and XII levels are decreased; factor V and factor VIII levels are preserved. Paradoxically, serum transaminase levels may be only modestly elevated; serum bilirubin concentration may be normal or only slightly elevated, in contrast to most forms of severe liver disease in which marked elevation of transaminases and serum bilirubin concentration occur concomitantly with prolongation of PT and PTT. Resistance of affected liver cells to cell death may explain the observed discrepancy in liver function [Vogel et al 2004].This early phase can progress to liver failure with ascites, jaundice, and gastrointestinal bleeding. Children may have a characteristic odor of "boiled cabbage" or "rotten mushrooms." Infants occasionally have persistent hypoglycemia; some have hyperinsulinism [Baumann et al 2005]. Others have chronic low-grade acidosis [CR Scott, unpublished data]. Untreated affected infants may die from liver failure within weeks or months of first symptoms [Hegarty et al 2015].
- Renal tubular involvement. In the more chronic form of the untreated disorder, symptoms develop after age six months; renal tubular involvement is the major manifestation. The renal tubular dysfunction involves a Fanconi-like renal syndrome with generalized aminoaciduria, phosphate loss, and, for many, renal tubular acidosis. The continued renal loss of phosphate is believed to account for rickets; serum calcium concentrations are usually normal.
- Neurologic crises. Untreated children may have repeated neurologic crises similar to those seen in older individuals with acute intermittent porphyria. These crises include change in mental status, abdominal pain, peripheral neuropathy, and/or respiratory failure requiring mechanical ventilation. Crises can last one to seven days. Repeated neurologic crises often go unrecognized:
- Mitchell et al [1990] reported that 42% of untreated French Canadian children with tyrosinemia type I had experienced such episodes.
- In an international survey, van Spronsen et al [1994] reported that 10% of deaths in untreated children occurred during a neurologic crisis.
- Hepatocellular carcinoma. Those children who are not treated with nitisinone and a low-tyrosine diet and who survive the acute onset of liver failure are at high risk of developing and succumbing to hepatocellular carcinoma.
- Survival in untreated children. Untreated infants diagnosed before age two months had a two-year survival rate of 29% [van Spronsen et al 1994].
- Those diagnosed between ages two and six months had a 74% two-year survival rate; those diagnosed after age six months had a 96% two-year survival rate.
- After more than five years the survival rate of the group diagnosed between ages two and six months dropped to approximately 30% and that of the group diagnosed after age six months dropped to approximately 60% (Figure 2).

Figure 2.
Survival of children with tyrosinemia before 1992 [van Spronsen et al 1994]
Treated tyrosinemia type I. The natural history in children who are treated with nitisinone is different from that in untreated children. Affected children younger than age two years who are treated with a combination of nitisinone and low-tyrosine diet are markedly improved compared to those children treated with low-tyrosine diet alone. The combined nitisinone and low-tyrosine diet treatment has resulted in a greater than 90% survival rate, normal growth, improved liver function, prevention of cirrhosis, correction of renal tubular acidosis, and improvement in secondary rickets [McKiernan 2006, Masurel-Paulet et al 2008, Larochelle et al 2012].
- Neurologic crises observed in treated children have always been associated with a prolonged interruption in nitisinone treatment [CR Scott, unpublished data].
- Children with acute liver failure require support prior to and during the initiation of treatment with nitisinone. Improvement generally occurs within one week of starting nitisinone treatment.
- Corneal crystals. Nitisinone blocks the tyrosine catabolic pathway such that succinylacetone is not produced but tissue tyrosine levels are raised. Blood tyrosine concentration greater than 600 mol/L confers risk of precipitation of tyrosine as bilateral, linear, branching subepithelial corneal opacities [Ahmad et al 2002], causing photophobia and itchy, sensitive eyes. The crystals resolve once tyrosine levels are reduced.
- Hepatocellular carcinoma. Although Holme & Lindstedt [2000] and van Spronsen et al [2005] reported hepatocellular carcinoma in individuals after years of nitisinone therapy, it is estimated that fewer than 5% of children placed on nitisinone therapy before age two years develop hepatocellular carcinoma by age ten years [Larochelle et al 2012].In Quebec, where tyrosinemia type I is included in the newborn screening program, no affected individuals have been hospitalized for manifestations of tyrosinemia type I, and hepatocellular carcinoma has not been reported in individuals who were placed on nitisinone therapy prior to age 30 days. The longest period of treatment reported in this group is 12 years [Larochelle et al 2012].
Pathophysiology
Fumarylacetoacetase (FAH) is the terminal enzyme in the tyrosine catabolic pathway (Figure 1). In FAH deficiency, fumarylacetoacetate (FAA), the immediate precursor:
- Appears to accumulate in hepatocytes, causing cellular damage and apoptosis (identified in an animal model by Endo & Sun [2002]);
- Is diverted into succinylacetoacetate and succinylacetone. Succinylacetone interferes with the activity of the following hepatic enzymes:
- Parahydroxyphenylpyruvic acid dioxygenase (p-HPPD), resulting in elevation of plasma tyrosine concentration;
- PBG synthase, resulting in (1) reduced activity of the enzyme δ-ALA dehydratase in liver and circulating red blood cells; (2) reduced heme synthesis; (3) increased δ-aminolevulinic acid (δ-ALA), which may induce acute neurologic episodes; and (4) increased urinary excretion of δ-ALA.
Genotype-Phenotype Correlations
In general, no correlation is observed between clinical presentation and genotype. Acute and chronic forms have been seen in the same families, as well as in unrelated individuals with the same genotype [Poudrier et al 1998].
One mechanism that explains this clinical variation is gene reversion. Hepatic nodules removed from livers of individuals with the chronic form of tyrosinemia type I have been shown to have cells that are immunologically positive for FAH protein and to have enzymatic activity for FAH [Kvittingen et al 1994, Grompe 2001]. These seemingly "normal" cells appear to have arisen by gene reversion – that is, the spontaneous self-correction (i.e., reversion or "back-mutation") of the germline pathogenic variant to the normal gene sequence during somatic cell division.
Spontaneous somatic variants that suppress the effects of the pathogenic variants and allow for normal or near-normal gene expression in these cells have also been reported [Bliksrud et al 2005]. This is a true reversion of the mutated sequence and not the result of maternal cell colonization or maternal cell fusion [Bergeron et al 2004]. The "normal" (i.e., reverted) cells have a selective growth advantage because they are no longer at risk for apoptosis from the accumulation of FAA. These foci of revertant "normal" cell colonies comprise many of the liver nodules in untreated individuals with chronic tyrosinemia type I who have a milder biochemical and clinical phenotype [Kim et al 2000, Demers et al 2003]. However, the continued production of FAA by the nonrevertant mutated cells places the individual at continued risk for hepatocellular carcinoma [Kim et al 2000].
A rare and atypical form of tyrosinemia type I has been reported in a four-month-old Belgian infant with severe liver disease. Liver function studies were abnormal with markedly elevated alpha-fetoprotein, prolonged PT and PTT, and undetectable succinylacetone in urine. Fumarylacetoacetase (FAH) protein and activity was decreased, but not absent. Homozygosity for a unique pathogenic variant, c.103G>A (p.Ala35Thr), was identified [Cassiman et al 2009].
Similarly, three sibs who developed chronic liver disease and HCC without detectable blood or urine succinylacetone had deficient FAH activity. The family was of Middle Eastern background and each child was homozygous for c.424A>G in FAH [Blackburn et al 2016].
Nomenclature
The term " tyrosinosis" was previously used to refer to tyrosinemia type I include tyrosinosis.
Prevalence
In geographic areas without newborn screening, tyrosinemia type I affects approximately one in 100,000 to 120,000 births [Mitchell et al 2001]. Because of the inconsistent and confusing nature of its clinical presentation, it is estimated that fewer than 50% of affected individuals are diagnosed while alive.
In the general US population, the carrier frequency is estimated at 1:150 to 1:100.
Two regions of the world have a higher than expected frequency of tyrosinemia type I due to the increased frequency of certain pathogenic variants resulting from a founder effect:
- In the Scandanavian countries (c.1062+5G>A (IVS 12+5 G>A), p.Gly337Ser, and/or p.249HisfsTer5.5) and in Finland (p.Trp262Ter), the birth prevalence is estimated at 1:74,000 and 1:60,000 live births, respectively [Bliksrud et al 2012].
- A founder effect from colonization by French settlers is present in the province of Quebec, Canada. The c.1062+5G>A (IVS12+5 G>A) pathogenic variant accounts for 87% of allelic variants in this population. The birth prevalence in the province of Quebec is 1:16,000. In the Saguenay-Lac Saint-Jean region of Quebec, it is 1:1,846 live births. The overall carrier frequency in Quebec is 1:66 based on newborn screening data. The carrier frequency in the Saguenay-Lac St-Jean region is 1:16-1:20.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with pathogenic variants in FAH.
Differential Diagnosis
Children with any of the following presenting findings (see Table 2) should be evaluated for tyrosinemia type I.
Table 2.
Differential Diagnosis of Tyrosinemia Type I in Infants by Presenting Finding
Tyrosinemia type II is caused by a defect in tyrosine aminotransferase (TAT) (EC 2.6.1.5). Establishing the diagnosis of tyrosinemia type II relies on the following:
- Plasma tyrosine concentration typically greater than 500 µmol/L that may exceed 1,000 µmol/L (The concentration of other amino acids is normal.)
- Increased excretion of p-hydroxyphenylpyruvate, p-hydroxyphenyllactate, and p-hydroxyphenylacetate and presence of small quantities of N-acetyltyrosine and 4-tyramine on urine organic acid analysis
Affected individuals have painful, nonpruritic, and hyperkeratotic plaques on the soles and palms. The plantar surface of the digits may show marked yellowish thickening associated with the hyperkeratosis. Ophthalmologic involvement is recalcitrant pseudodendritic keratitis [Macsai et al 2001]. Although developmental delay appears to be common, it is unclear if ascertainment bias accounts for this and the reports of neurologic symptoms.
Findings improve on a diet restricted in tyrosine and phenylalanine [Ellaway et al 2001].
Tyrosinemia type III, the rarest of the tyrosine disorders, is caused by a deficiency of p-hydroxyphenylpyruvic acid dioxygenase (EC.1.13.11.27). Plasma concentration of tyrosine ranges from 350 to 650 µmol/L. Excretion of 4-hydroxyphenylpyruvic acid, 4-hydroxyphenyllactate, and 4-hydroxyphenylacetate is increased. The precise quantities vary with protein intake.
Few individuals with the disorder have been identified, and the clinical phenotype remains ill defined. The first affected individuals came to medical attention because of intellectual disability or ataxia; another was detected on routine screening [Mitchell et al 2001]. These individuals, like those with tyrosinemia type II, have no liver involvement but have skin or ocular changes. It remains unclear if tyrosinemia type III is truly associated with cognitive delays or if the association has resulted from ascertainment bias [Ellaway et al 2001].
A diet low in phenylalanine and tyrosine can lower plasma tyrosine concentration.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of a child diagnosed with tyrosinemia type I on the basis of newborn screening, the following evaluations are recommended (see Table 3):
- CBC with platelet count
- Serum concentration of electrolytes
- Assessment of liver function (PT, PTT, AST, ALT, GGT, serum bilirubin concentration, alkaline phosphatase, and serum AFP).
For children ascertained on the basis of clinical symptoms (see Table 4; pdf), testing should include the evaluations listed above in addition to:
- Baseline abdominal imagining by CT or MRI (with contrast) to evaluate for liver adenomas or nodules (see Dubois et al [1996]) and renal size;
- X-ray of wrist to document presence or absence of rickets.
Clinical genetics consultation is indicated for all affected individuals.
Treatment of Manifestations
Management guidelines have been published. US recommendations include Chinsky et al [2017]; European recommendations include de Laet et al [2013].
Acute management of liver failure. Children may require respiratory support, appropriate fluid management, and blood products for correction of bleeding diathesis.
Nitisinone (Orfadin®). 2-(2-nitro-4-trifluoro-methylbenzyol) 1,3 cyclohexanedione (NTBC) blocks parahydroxyphenylpyruvic acid dioxygenase (p-HPPD), the second step in the tyrosine degradation pathway, and prevents the accumulation of FAA and its conversion to succinylacetone (Figure 1).
- Nitisinone should be prescribed as soon as the diagnosis of tyrosinemia type I is confirmed.
- Nitisinone is generally prescribed at 1.0 mg/kg/day; individual requirements may vary. Dosage should be adjusted to maintain blood nitisinone levels between 40 and 60 µmol/L, which theoretically blocks more than 99% of p-HPPD activity. Rarely, an individual may require higher blood levels of nitisinone (70 µmol/L) to suppress succinylacetone excretion. As long as blood concentration of nitisinone is within the therapeutic range, urine succinylacetone does not need to be measured.
- Nitisinone is typically given in two divided doses; however, because of the long half-life (50-60 hours), affected individuals who are older than one year and stable may maintain adequate therapy with 1x/day dosing [Schlune et al 2012].
- Rare side effects of nitisinone have included: transient low platelet count and transient low neutrophil count that resolved without intervention; and photophobia that resolved with stricter dietary control and subsequent lowering of blood tyrosine concentrations.
Low-tyrosine diet. Nitisinone increases blood concentration of tyrosine, necessitating a low-tyrosine diet to prevent tyrosine crystals from forming in the cornea.
- Dietary management should be started immediately upon diagnosis and should provide a nutritionally complete diet with controlled intakes of phenylalanine and tyrosine using a vegetarian diet with low-protein foods and a medical formula such as Tyrex® (Ross) or Tyros-1® (Mead Johnson).
- Phenylalanine and tyrosine requirements are interdependent and vary from individual to individual and within the same individual depending on growth rate, adequacy of energy and protein intakes, and state of health. With appropriate dietary management, plasma tyrosine concentration should be 300-600 µmol/L, regardless of age; plasma phenylalanine concentration should be 20-80 µmol/L (0.3-1.3 mg/dL). If the blood concentration of phenylalanine is too low (<20 µmol/L), additional protein should be added to the diet from milk or foods.
Liver transplantation. Prior to the availability of nitisinone for the treatment of tyrosinemia type I, the only definitive therapy was liver transplantation.
- Liver transplantation should be reserved for those children who (1) have severe liver failure at clinical presentation and fail to respond to nitisinone therapy or (2) have documented evidence of malignant changes in hepatic tissue [Bartlett et al 2014].
- Transplant recipients require long-term immunosuppression. Mortality associated with liver transplantation in young children is approximately 10%.
- Transplant recipients may also benefit from low-dose (0.1mg/kg/day) nitisinone therapy to prevent continued renal tubular and glomerular dysfunction resulting from persistence of succinylacetone in the plasma and urine [Bartlett et al 2013].
Prevention of Primary Manifestations
Treatment with nitisinone (Orfadin®) should begin as soon as the diagnosis is confirmed.
Prevention of Secondary Complications
Because carnitine deficiency secondary to the renal tubular Fanconi syndrome can cause skeletal muscle weakness, serum concentration of carnitine should be measured so that carnitine deficiency, if identified, can be treated [Nissenkorn et al 2001].
Osteoporosis and rickets resulting from renal tubular damage are treated by correction of acidosis, restoring of calcium and phosphate balance, and administration of 25-hydroxy-vitamin D.
Surveillance
Frequent evaluation of the following parameters is typical in the management of individuals with tyrosinemia type I (Table 3) [CR Scott, personal recommendations].
Table 3.
Suggested Guidelines for Monitoring in Individuals with Tyrosinemia Type I Diagnosed by Newborn Screening
For monitoring children diagnosed based on clinical presention, see Table 4 (pdf).
For alternative management recommendations, see de Laet et al [2013].
Agents/Circumstances to Avoid
Avoid inappropriate protein intake.
Evaluation of Relatives at Risk
It is appropriate to evaluate apparently asymptomatic older and younger sibs of a proband in order to identify as early as possible those who would benefit from prompt initiation of treatment and preventive measures. Evaluations can include:
- Molecular genetic testing if the pathogenic variants in the family are known. Prenatal testing can be used to clarify the genetic status of at-risk sibs before birth.
- Blood or urine succinylacetone analysis as soon as possible after birth if prenatal testing was not performed. This allows prompt initiation of treatment, as postnatal genetic testing results may not be available in a timely fashion.
- If the pathogenic variants in the family are not known, consideration of analysis for urine succinylacetone in healthy older sibs
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Little data exist on the use of nitisinone during human pregnancy. Speculation would assume that the pregnant woman remains safe from untoward events; however, the developing fetus may be at risk because of alterations in tyrosine metabolism.
At least two women have given birth to healthy infants while receiving therapeutic doses of nitisinone [Garcia Segarra et al 2010].
- In one instance the affected mother gave birth to an unaffected infant who is reported to be healthy and developing normally at age 2.5 years [Garcia Segarra et al 2010, Vanclooster et al 2012].
- In another instance, an affected woman gave birth to an affected child. The child is reported to have normal growth and development at age seven months [Garcia Segarra et al 2010]. The authors speculate that the affected child was protected from in utero liver damage by maternal treatment with nitisinone during pregnancy.
See MotherToBaby for more information on medication exposure during pregnancy.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Other
Prior to the availability of nitisinone, the only available non-transplant therapy was a diet limiting the availability of phenylalorine and tyrosine. Although the diet was modestly helpful, recurrent episodes of neurologic crises and progression of liver disease occurred. Average survival was less than age ten years.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Tyrosinemia type I is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., carriers of one FAH pathogenic variant).
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. The offspring of an individual with tyrosinemia type I are obligate heterozygotes (carriers) for a pathogenic variant in FAH.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a FAH pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the FAH pathogenic variants in the family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, allelic variants, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative genetic alteration/s are unknown).
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the FAH pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for tyrosinemia type I are possible.
Biochemical testing. Prenatal testing for pregnancies at 25% risk is possible by detection of succinylacetone in amniotic fluid obtained by amniocentesis usually performed at approximately 15 to 18 weeks' gestation. Although detection of succinylacetone in amniotic fluid is diagnostic, false negative results have been reported; thus, this method should only be used by laboratories consistently able to identify succinylacetone at low levels by stable isotope detection. Because of these issues with biochemical testing, molecular genetic testing is the preferred method of prenatal diagnosis.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- British Inherited Metabolic Disease Group (BIMDG)TEMPLE (Tools Enabling Metabolic Parents LEarning)United Kingdom
- Medical Home Portal
- MedlinePlus
- American Liver FoundationPhone: 800-465-4837 (HelpLine)
- Metabolic Support UKUnited KingdomPhone: 0845 241 2173
- Newborn Screening in Your StateHealth Resources & Services Administration
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Tyrosinemia Type I: Genes and Databases
Table B.
OMIM Entries for Tyrosinemia Type I (View All in OMIM)
Gene structure. FAH is approximately 35 kbp in size and comprises 14 exons. For a detailed summary of gene and protein information, see Table A, Gene.
Benign variants. A single pseudodeficiency allele, c.1021C>T (p.Arg341Trp), leads to decreased FAH enzyme activity and very little immunoreactive protein, but adequate amounts of FAH mRNA [Rootwelt et al 1994].
Pathogenic variants. See Table 5. Pathogenic missense, nonsense, and splice site variants, as well as small deletions and indels of FAH, have been reported. Park et al [2009] reported a large deletion involving FAH.
The following population-specific pathogenic variants result from founder effects or genetic drift [Angileri et al 2015] (see Table 5):
- Ashkenazi Jewish: p.Pro261Leu
- Finnish: p.Trp262Ter
- French Canadian: c.1062+5G>A
- Pakistani: p.Gln64His
- Scandinavian: p.Gly337Ser
- Turkish: p.Asp233Val
- Northern European: c.1062+5G>A
- Southern European: c.554-1G>T
Table 5.
FAH Variants Discussed in This GeneReview
For additional pathogenic variants identified in North American patients, see Table 6 (pdf).
Normal gene product. FAH is a cytosolic protein that acts as a homodimer and has a molecular weight of approximately 80 kd. The wild type FAH has a Km for FAA of about 3.5 μmol/L. FAH catalyzes the conversion of FAA to fumarate and acetoacetate and the conversion of succinylacetoacetate to succinate and acetoacetate.
Abnormal gene product. Pathogenic missense, nonsense, and splice site variants result in loss of FAH enzyme activity, leading to an intracellular accumulation of FAA resulting in cellular damage and apoptosis.
References
Literature Cited
- Ahmad S, Teckman JH, Lueder GT. Corneal opacities associated with NTBC treatment. Am J Ophthalmol. 2002;134:266–8. [PubMed: 12140036]
- Angileri F, Bergeron A, Morrow G, Lettre F, Gray G, Hutchin T, Ball S, Tanguay RM. Geographical and ethnic distribution of mutations of the fumarylacetoacetate hydrolase gene in hereditary tyrosinemia type 1. JIMD Rep. 2015;19:43–58. [PMC free article: PMC4501228] [PubMed: 25681080]
- Bansal S, Dhawan A. Acute liver failure. In: Walker WA, Kleinman RE, Sherman PM, Shneider BL, Sanderson IR, eds. Pediatric Gastrointestinal Disease - Pathophysiology, Diagnosis, Management. 4 ed. Ch 58. Hamilton, Ontario: BC Decker, Inc; 2004:1491-507.
- Bartlett DC, Lloyd C, McKiernan PJ, Newsome PN. Early nitisinone treatment reduces the need for liver transplantation in children with tyrosinaemia type 1 and improves post-transplant renal function. J Inherit Metab Dis. 2014;37:745–52. [PubMed: 24515874]
- Bartlett DC, Preece MA, Holme E, Lloyd C, Newsome PN, McKiernan PJ. Plasma succinylacetone is persistently raised after liver transplantation in tyrosinaemia type 1. J Inherit Metab Dis. 2013;36:15–20. [PubMed: 22456946]
- Baumann U, Preece MA, Green A, Kelly DA, McKiernan PJ. Hyperinsulinism in tyrosinaemia type I. J Inherit Metab Dis. 2005;28:131–5. [PubMed: 15877201]
- Bergeron A, Lettre F, Russo P, Morissette J, Tanguay RM. No evidence of maternal cell colonization in reverted liver nodules of tyrosinemia type I patients. Gastroenterology. 2004;127:1381–5. [PubMed: 15521007]
- Blackburn PR, Hickey RD, Nace RA, Giama NH, Kraft DL, Bordner AJ, Chaiteerakij R, McCormick JB, Radulovic M, Graham RP, Torbenson MS, Tortorelli S, Scott CR, Lindor NM, Milliner DS, Oglesbee D, Al-Qabandi W, Grompe M, Gavrilov DK, El-Youssef M, Clark KJ, Atwal PS, Roberts LR, Klee EW, Ekker SC. Silent tyrosinemia type I without elevated tyrosine or succinylacetone associated with liver cirrhosis and hepatocellular carcinoma. Hum Mutat. 2016;37:1097–105. [PMC free article: PMC5108417] [PubMed: 27397503]
- Bliksrud YT, Brodtkorb E, Backe PH, Woldseth B, Rootwelt H. Hereditary tyrosinaemia type I in Norway: incidence and three novel small deletions in the fumarylacetoacetase gene. Scand J Clin Lab Invest. 2012;2012;72:369–73. [PubMed: 22554029]
- Bliksrud YT, Brodtkorb E, Andresen PA, van den Berg IE, Kvittingen EA. Tyrosinaemia type I--de novo mutation in liver tissue suppressing an inborn splicing defect. J Mol Med. 2005;83:406–10. [PubMed: 15759101]
- Cassiman D, Zeevaert R, Holme E, Kvittingen EA, Jaeken J. A novel mutation causing mild, atypical fumarylacetoacetase deficiency (Tyrosinemia type I): a case report. Orphanet J Rare Dis. 2009;4:28. [PMC free article: PMC2802351] [PubMed: 20003495]
- Chinsky JM, Singh R, Ficicioglu C, van Karnebeek CDM, Grompe M, Mitchell G, Waisbren SE, Gucsavas-Calikoglu M, Wasserstein MP, Coakley K, Scott CR. Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations. Genet Med. 2017;19(12) [PMC free article: PMC5729346] [PubMed: 28771246]
- Demers SI, Russo P, Lettre F, Tanguay RM. Frequent mutation reversion inversely correlates with clinical severity in a genetic liver disease, hereditary tyrosinemia. Hum Pathol. 2003;34:1313–20. [PubMed: 14691918]
- Dubois J, Garel L, Patriquin H, Paradis K, Forget S, Filiatrault D, Grignon A, Russo P, St-Vil D. Imaging features of type 1 hereditary tyrosinemia: a review of 30 patients. Pediatr Radiol. 1996;26:845–51. [PubMed: 8929295]
- Ellaway CJ, Holme E, Standing S, Preece MA, Green A, Ploechl E, Ugarte M, Trefz FK, Leonard JV. Outcome of tyrosinaemia type III. J Inherit Metab Dis. 2001;24:824–32. [PubMed: 11916315]
- Elpeleg ON, Shaag A, Holme E, Zughayar G, Ronen S, Fisher D, Hurvitz H. Mutation analysis of the FAH gene in Israeli patients with tyrosinemia type I. Hum Mutat. 2002;19:80–1. [PubMed: 11754109]
- Endo F, Sun MS. Tyrosinaemia type I and apoptosis of hepatocytes and renal tubular cells. J Inherit Metab Dis. 2002;25:227–34. [PubMed: 12137232]
- Garcia Segarra N, Roche S, Imbard A, Benoist JF, Grenèche MO, Davit-Spraul A, Ogier de Baulny H. Maternal and fetal tyrosinemia type I. J Inherit Metab Dis. 2010;33 Suppl 3:S507–10. [PubMed: 23250512]
- Giguère Y, Ruel J, Belanger N, Grenier A, Laberge C, Quebec Neonatal Blood Screening Programme, CHUL du CHUQ, Quebec, Canada. Neonatal mass screening for hereditary tyrosinemia type 1 in Quebec: a historical perspective (1970-2005). Portland, OR: Newborn Screening and Genetic Testing Symposium. 2005.
- Grompe M. The pathophysiology and treatment of hereditary tyrosinemia type 1. Semin Liver Dis. 2001;21:563–71. [PubMed: 11745044]
- Hegarty R, Hadzic N, Gissen P, Dhawan A. Inherited metabolic disorders presenting as acute liver failure in newborns and young children: King's College Hospital experience. Eur J Pediatr. 2015;174:1387–92. [PubMed: 25902754]
- Hendriksz CJ, Walter JH. Feeding infants with undiluted goat's milk can mimic tyrosinaemia type 1. Acta Paediatr. 2004;93:552–3. [PubMed: 15188986]
- Holme E, Lindstedt S. Nontransplant treatment of tyrosinemia. Clin Liver Dis. 2000;4:805–14. [PubMed: 11232358]
- Kim SZ, Kupke KG, Ierardi-Curto L, Holme E, Greter J, Tanguay RM, Poudrier J, D'Astous M, Lettre F, Hahn SH, Levy HL. Hepatocellular carcinoma despite long-term survival in chronic tyrosinaemia I. J Inherit Metab Dis. 2000;23:791–804. [PubMed: 11196105]
- Kvittingen EA, Rootwelt H, Berger R, Brandtzaeg P. Self-induced correction of the genetic defect in tyrosinemia type I. J Clin Invest. 1994;94:1657–61. [PMC free article: PMC295327] [PubMed: 7929843]
- de Laet C, Dionisi-Vici C, Leonard JV, McKiernan P, Mitchell G, Monti L, de Baulny HO, Pintos-Morell G, Spiekerkötter U. Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis. 2013;2013;8:8. [PMC free article: PMC3558375] [PubMed: 23311542]
- Larochelle J, Alvarez F, Bussières JF, Chevalier I, Dallaire L, Dubois J, Faucher F, Fenyves D, Goodyer P, Grenier A, Holme E, Laframboise R, Lambert M, Lindstedt S, Maranda B, Melançon S, Merouani A, Mitchell J, Parizeault G, Pelletier L, Phan V, Rinaldo P, Scott CR, Scriver C, Mitchell GA. Effect of nitisinone (NTBC) treatment on the clinical course of hepatorenal tyrosinemia in Québec. Mol Genet Metab. 2012;107:49–54. [PubMed: 22885033]
- Macsai MS, Schwartz TL, Hinkle D, Hummel MB, Mulhern MG, Rootman D. Tyrosinemia type II: nine cases of ocular signs and symptoms. Am J Ophthalmol. 2001;132:522–7. [PubMed: 11589874]
- Masurel-Paulet A, Poggi-Bach J, Rolland MO, Bernard O, Guffon N, Dobbelaere D, Sarles J, de Baulny HO, Touati G. NTBC treatment in tyrosinaemia type 1: long-term outcome in French patients. J Inherit Metab Dis. 2008;31:81–7. [PubMed: 18214711]
- McKiernan PJ. Nitisinone in the treatment of hereditary tyrosinaemia type 1. Drugs. 2006;66:743–50. [PubMed: 16706549]
- Mitchell G, Larochelle J, Lambert M, Michaud J, Grenier A, Ogier H, Gauthier M, Lacroix J, Vanasse M, Larbrisseau A, et al. Neurologic crises in hereditary tyrosinemia. N Engl J Med. 1990;322:432–7. [PubMed: 2153931]
- Mitchell GA, Grompe M, Lambert M, Tanguay RM. Hypertyrosinemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw Hill; 2001:1777-806.
- Nissenkorn A, Korman SH, Vardi O, Levine A, Katzir Z, Ballin A, Lerman-Sagie T. Carnitine-deficient myopathy as a presentation of tyrosinemia type I. J Child Neurol. 2001;16:642–4. [PubMed: 11575602]
- Park HD, Lee DH, Choi TY, Lee YK, Kim JW, Ki CS, Lee YW. Clinical, biochemical, and genetic analysis of a Korean neonate with hereditary tyrosinemia type 1. Clin Chem Lab Med. 2009;47:930–3. [PubMed: 19569981]
- Poudrier J, Lettre F, Scriver CR, Larochelle J, Tanguay RM. Different clinical forms of hereditary tyrosinemia (type I) in patients with identical genotypes. Mol Genet Metab. 1998;64:119–25. [PubMed: 9705236]
- Poudrier J, St-Louis M, Lettre F, Gibson K, Prevost C, Larochelle J, Tanguay RM. Frequency of the IVS12 + 5G-->A splice mutation of the fumarylacetoacetate hydrolase gene in carriers of hereditary tyrosinaemia in the French Canadian population of Saguenay-Lac-St-Jean. Prenat Diagn. 1996;16:59–64. [PubMed: 8821854]
- Rootwelt H, Brodtkorb E, Kvittingen EA. Identification of a frequent pseudodeficiency mutation in the fumarylacetoacetase gene, with implications for diagnosis of tyrosinemia type I. Am J Hum Genet. 1994;55:1122-7. [PMC free article: PMC1918441] [PubMed: 7977370]
- Sassa S, Kappas A. Hereditary tyrosinemia and the heme biosynthetic pathway. Profound inhibition of delta-aminolevulinic acid dehydratase activity by succinylacetone. J Clin Invest. 1983;71:625–34. [PMC free article: PMC436912] [PubMed: 6826727]
- Schlune A, Thimm E, Herebian D, Spiekerkoetter U. Single dose NTBC-treatment of hereditary tyrosinemia type I. J Inherit Metab Dis. 2012;2012;35:831–6. [PubMed: 22307209]
- Schulze A, Frommhold D, Hoffmann GF, Mayatepek E. Spectrophotometric microassay for delta-aminolevulinate dehydratase in dried-blood spots as confirmation for hereditary tyrosinemia type I. Clin Chem. 2001;47:1424–9. [PubMed: 11468232]
- Techakittiroj C, Cunningham A, Hooper PF, Andersson HC, Thoene J. High protein diet mimics hypertyrosinemia in newborn infants. J Pediatr. 2005;146:281–2. [PubMed: 15689925]
- Vanclooster A, Devlieger R, Meersseman W, Spraul A, Kerckhove KV, Vermeersch P, Meulemans A, Allegaert K, Cassiman D. Pregnancy during nitisinone treatment for tyrosinaemia type I: first human experience. JIMD Rep. 2012;5:27–33. [PMC free article: PMC3509920] [PubMed: 23430914]
- van Spronsen FJ, Bijleveld CM, van Maldegem BT, Wijburg FA. Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4-3 trifl). J Pediatr Gastroenterol Nutr. 2005;40:90–3. [PubMed: 15625434]
- van Spronsen FJ, Thomasse Y, Smit GP, Leonard JV, Clayton PT, Fidler V, Berger R, Heymans HS. Hereditary tyrosinemia type I: a new clinical classification with difference in prognosis on dietary treatment. Hepatology. 1994;20:1187–91. [PubMed: 7927251]
- Vogel A, van Den Berg IE, Al-Dhalimy M, Groopman J, Ou CN, Ryabinina O, Iordanov MS, Finegold M, Grompe M. Chronic liver disease in murine hereditary tyrosinemia type 1 induces resistance to cell death. Hepatology. 2004;39:433–43. [PubMed: 14767996]
Chapter Notes
Acknowledgments
Supported by grants from the Food and Drug Administration (FD-4-001445) and Rare Disease Therapeutics. The authors are appreciative of the collaboration and discussions with Dr Grant Mitchell of Montreal, Canada, and Dr Sven Lindstedt and Dr Elisabeth Holme of Gothenburg, Sweden.
Revision History
- 25 May 2017 (ma) Comprehensive update posted live
- 17 July 2014 (me) Comprehensive update posted live
- 25 August 2011 (me) Comprehensive update posted live
- 21 October 2008 (cg) Comprehensive update posted live
- 24 July 2006 (me) Review posted live
- 29 June 2005 (crs) Original submission
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: July 24, 2006; Last Update: May 25, 2017.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Sniderman King L, Trahms C, Scott CR. Tyrosinemia Type I. 2006 Jul 24 [Updated 2017 May 25]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.