Summary
Clinical characteristics.
VLDLR cerebellar hypoplasia (VLDLR-CH) is characterized by non-progressive congenital ataxia that is predominantly truncal and results in delayed ambulation, moderate-to-profound intellectual disability, dysarthria, strabismus, and seizures. Children either learn to walk very late (often after age 6 years) or never achieve independent ambulation. Brain MRI findings include hypoplasia of the inferior portion of the cerebellar vermis and hemispheres, simplified gyration of the cerebral hemispheres, and small brain stem – particularly the pons.
Diagnosis/testing.
The diagnosis of VLDLR cerebellar hypoplasia is established in a proband with suggestive clinical and brain MRI findings by identification of biallelic pathogenic variants in VLDLR on molecular genetic testing.
Management.
Treatment of manifestations: Seizures and strabismus are treated in the standard manner. Referral to an early intervention program is recommended for access to occupational, physical, and speech therapy, as well as infant mental health services and special educators.
Surveillance: Annual neurologic and rehabilitation evaluations.
Genetic counseling.
VLDLR-CH is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible when the pathogenic variants in a family are known.
Diagnosis
VLDLR cerebellar hypoplasia (VLDLR-CH) is a subgroup of dysequilibrium syndrome (DES), a spectrum of genetically heterogeneous conditions that combines non-progressive cerebellar ataxia with intellectual disability inherited in an autosomal recessive manner.
Suggestive Findings
VLDLR cerebellar hypoplasia should be suspected in individuals with the following major diagnostic features:
- Non-progressive congenital ataxia that is predominantly truncal and results in delayed ambulation
- Moderate-to-profound intellectual disability
- Dysarthria
- MRI findings (see Figure 1) that include the following:
- Hypoplasia of the inferior portion of the cerebellar vermis and hemispheres
- Simplified gyration of the cerebral hemispheres with minimally thickened but uniform cortex and lack of clear anteroposterior gradient
- Small brain stem, particularly the pons
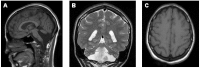
Figure 1.
MRI of the brain demonstrating typical neuroimaging findings of VLDLR-CH A. Sagittal T1-weighted
Establishing the Diagnosis
The diagnosis of VLDLR cerebellar hypoplasia is established in a proband by identification of biallelic pathogenic (or likely pathogenic) variants in VLDLR on molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of biallelic VLDLR variants of uncertain significance (or of one known VLDLR pathogenic variant and one VLDLR variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (chromosomal microarray analysis, exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of VLDLR cerebellar hypoplasia is often recognizable, individuals with the distinctive MRI findings described in Suggestive Findings can often be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of VLDLR cerebellar hypoplasia has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and radiographic findings suggest the diagnosis of VLDLR cerebellar hypoplasia, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of VLDLR is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.Note: Targeted analysis for pathogenic variants can be performed first in individuals of Hutterite ancestry. See Table 6.
- A multigene panel that includes VLDLR and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis can be considered if only one or no pathogenic variant is found on sequencing.
Option 2
When the diagnosis of VLDLR cerebellar hypoplasia is not considered because of its rarity and/or because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in VLDLR Cerebellar Hypoplasia
Clinical Characteristics
Clinical Description
VLDLR cerebellar hypoplasia is a congenital non-progressive disorder characterized by cerebellar ataxia and intellectual disability.
To date, more than 50 individuals have been identified with a pathogenic variant in VLDLR [Boycott et al 2005, Glass et al 2005, Moheb et al 2008, Ozcelik et al 2008, Türkmen et al 2008, Boycott et al 2009, Kolb et al 2010, Ali et al 2012, Azmanov et al 2013, Kruer et al 2013, Schlotawa et al 2013, Sonmez et al 2013, Giorgio et al 2016, Micalizzi et al 2016, Valence et al 2016, Wilker et al 2019]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Features of VLDLR Cerebellar Hypoplasia
Brain MRI. All affected individuals demonstrate hypoplasia of the inferior portion of the cerebellar vermis and hemispheres. In addition, the majority of affected individuals show a simplified gyration of the cerebral hemispheres with minimally thickened but uniform cortex, lack of clear anteroposterior gradient, and small brain stem (particularly the pons). Some individuals are described as demonstrating neuroimaging features of pontocerebellar hypoplasia.
Cerebellar ataxia. All affected individuals demonstrate significant truncal ataxia. Children either learn to walk very late (often after age 6 years) or never achieve independent ambulation. For those able to ambulate independently, gait is wide based; affected individuals are not able to perform a tandem gait. Affected individuals from Turkey demonstrate quadrupedal locomotion in which the palms of the hands touch the ground and the elbows, back, and knees are straight [Ozcelik et al 2008, Türkmen et al 2009], an interesting behavioral adaptation which likely depends on the presence of special environmental influences during child development [Herz et al 2008, Türkmen et al 2009]. Limb ataxia is present in most individuals but is not severe.
Intellectual disability. All reported affected individuals have intellectual disability, ranging from moderate to profound. Most individuals can follow simple commands. Some can communicate verbally using short phrases or sentences. Adults are unable to live independently.
Dysarthria. Those who are able to communicate verbally demonstrate dysarthria.
Strabismus. The majority of individuals have strabismus.
Other
- Nystagmus is reported in some individuals and is described as gaze evoked.
- Epileptic seizures were reported in 40% of the affected individuals from the Hutterite population [Glass et al 2005], and appear to be less frequent in non-Hutterite individuals. The seizures tend to be generalized.
- Deep tendon reflexes in the lower extremities tend to be brisk.
- Microcephaly (2-4 SD below the mean) has been reported in a few affected individuals.
- Short stature (height just below the 2nd centile) is a feature in a few affected individuals.
Life span. There has been no formal study of life span in this disorder, but experience from the Hutterite population suggests that life span is not significantly reduced.
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been identified.
Nomenclature
VLDLR-CH is a clinically and molecularly well-defined subgroup of dysequilibrium syndrome (DES).
Prevalence
The actual frequency of VLDLR-CH is unknown.
More than 25 individuals with VLDLR-CH from the Hutterite population in Canada and the US have been followed for many years. This condition is present in all three Hutterite leuts (branches) (i.e., Schmiedeleut, Lehrerleut, and Dariusleut).
The estimated carrier frequency in the Hutterite population is one in 15 [Glass et al 2005].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in VLDLR.
Differential Diagnosis
The differential diagnosis of VLDLR cerebellar hypoplasia (VLDLR-CH) includes autosomal recessive conditions characterized by congenital or very early-onset cerebellar ataxia associated with cerebellar hypoplasia. Because cerebellar hypoplasia can be difficult to distinguish from cerebellar atrophy on early imaging, conditions characterized by the latter should also be considered (see Table 3).
Note: Diverse phenotypes associated with childhood- and adult-onset ataxia are to be excluded (see Hereditary Ataxia Overview, Table 3. Autosomal Recessive Cerebellar Ataxias: Single-Gene Disorders).
Table 3.
Genes and Disorders of Interest in the Differential Diagnosis of VLDLR Cerebellar Hypoplasia
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with VLDLR cerebellar hypoplasia (VLDLR-CH), the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with VLDLR Cerebellar Hypoplasia
Treatment of Manifestations
Treatment of seizures and strabismus is done in the standard manner.
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- As required by special education law, children should be in the least restrictive environment feasible at school and included in general education as much as possible and when appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to promote ambulation.
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Communication issues. Consider evaluation for alternative means of communication (e.g., Augmentative and Alternative Communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, and in many cases can improve it.
Surveillance
Table 5.
Recommended Surveillance for Individuals with VLDLR Cerebellar Hypoplasia
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
VLDLR cerebellar hypoplasia (VLDLR-CH) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., carriers of one VLDLR pathogenic variant).
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. To date, individuals with VLDLR-CH are not known to reproduce.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a VLDLR pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the VLDLR pathogenic variants in the family.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are carriers or are at risk of being carriers.
Population screening. In the Hutterite population, the high carrier frequency (~1:15) could warrant population screening for reproductive purposes [Glass et al 2005]. Carrier testing for the Hutterite population involves targeted analysis for the founder deletion [Boycott et al 2005].
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Once the VLDLR pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing for VLDLR-CH are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- National Institute of Neurological Disorders and Stroke (NINDS)PO Box 5801Bethesda MD 20824Phone: 800-352-9424 (toll-free); 301-496-5751; 301-468-5981 (TTY)
- American Association on Intellectual and Developmental Disabilities (AAIDD)Phone: 202-387-1968
- CDC - Child DevelopmentPhone: 800-232-4636
- National Ataxia FoundationPhone: 763-553-0020Email: naf@ataxia.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
VLDLR Cerebellar Hypoplasia: Genes and Databases
Table B.
OMIM Entries for VLDLR Cerebellar Hypoplasia (View All in OMIM)
Molecular Pathogenesis
VLDLR encodes a protein of 873 amino acids and is expressed abundantly in the heart, skeletal muscle, kidney, and brain. VLDLR protein is part of the reelin signaling pathway, which guides neuroblast migration in the developing cerebral cortex and cerebellum [Tissir & Goffinet 2003]. In an evolutionarily conserved pathway, reelin engages two lipoprotein receptors, VLDLR and apolipoprotein E receptor-2 (Apoer2), resulting in phosphorylation of disabled-1 (Dab1) and activation of an intracellular signaling cascade that allows neuroblasts to complete migration.
VLDLR belongs to a subset of cell surface receptors called the LDL receptor protein family. Family members share a number of domains arranged in a similar pattern: ligand-binding repeat domain, EGF repeat, YWTD domain, O-linked sugar domain, transmembrane domain, and a cytoplasmic domain containing a NPXY motif. VLDLR was initially identified to function in the receptor-mediated endocytosis of apoE-containing lipoproteins.
Mechanism of disease causation. All of the reported pathogenic variants to date are predicted to result in loss of function of the VLDLR protein. In the absence of this receptor, neuroblasts are unable to complete migration and adopt their ultimate position in the developing central nervous system.
Table 6.
Notable VLDLR Pathogenic Variants
Chapter Notes
Revision History
- 27 February 2020 (ha) Comprehensive update posted live
- 8 August 2013 (me) Comprehensive update posted live
- 26 August 2008 (cg) Review posted live
- 7 July 2008 (kmb) Original submission
References
Literature Cited
- Ali BR, Silhavy JL, Gleeson MJ, Gleeson JG, Al-Gazali L. A missense founder mutation in VLDLR is associated with dysequilibrium syndrome without quadrupedal locomotion. BMC Med Genet. 2012;13:80. [PMC free article: PMC3495048] [PubMed: 22973972]
- Azmanov DN, Chamova T, Tankard R, Gelev V, Bynevelt M, Florez L, Tzoneva D, Zlatareva D, Guergueltcheva V, Bahlo M, Tournev I, Kalaydjieva L. Challenges of diagnostic exome sequencing in an inbred founder population. Mol Genet Genomic Med. 2013;1:71-6. [PMC free article: PMC3865571] [PubMed: 24498604]
- Boycott KM, Bonnemann C, Herz J, Neuert S, Beaulieu C, Scott JN, Venkatasubramanian A, Parboosingh JS. Mutations in VLDLR as a cause for autosomal recessive cerebellar ataxia with mental retardation (dysequilibrium syndrome). J Child Neurol. 2009;24:1310-5. [PMC free article: PMC2849979] [PubMed: 19332571]
- Boycott KM, Flavelle S, Bureau A, Glass HC, Fujiwara TM, Wirrell E, Davey K, Chudley AE, Scott JN, McLeod DR, Parboosingh JS. Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am J Hum Genet. 2005;77:477-83. [PMC free article: PMC1226212] [PubMed: 16080122]
- Giorgio E, Ciolfi A, Biamino E, Caputo V, Di Gregorio E, Belligni EF, Calcia A, Gaidolfi E, Bruselles A, Mancini C, Cavalieri S, Molinatto C, Cirillo Silengo M, Ferrero GB, Tartaglia M, Brusco A. Whole exome sequencing is necessary to clarify ID/DD cases with de novo copy number variants of uncertain significance: Two proof-of-concept examples. Am J Med Genet A. 2016;170:1772-9. [PubMed: 27108886]
- Glass HC, Boycott K, Adams C, Barlow K, Scott JN, Chudley AE, Fujiwara T, Morgan K, Wirrell E, McLeod D. Autosomal recessive cerebellar hypoplasia in the Hutterite population: a syndrome of nonprogressive cerebellar ataxia with mental retardation. Dev Med Child Neurol. 2005;47:691-5. [PubMed: 16174313]
- Herz J, Boycott KM, Parboosingh JS. "Devolution" of bipedality. Proc Natl Acad Sci U S A. 2008;105:E25. [PMC free article: PMC2396673] [PubMed: 18487453]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389-97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Kolb LE, Arlier Z, Yalcinkaya C, Ozturk AK, Moliterno JA, Erturk O, Bayrakli F, Korkmaz B, DiLuna ML, Yasuno K, Bilguvar K, Ozcelik T, Tuysuz B, State MW, Gunel M. Novel VLDLR microdeletion identified in two Turkish siblings with pachygyria and pontocerebellar atrophy.Neurogenetics. 2010;11:319-25. [PubMed: 20082205]
- Kruer MC, Jepperson TN, Weimer JM, Mroch A, Davis-Keppen L, Crotwell P, Parboosingh J. Mutations in VLDLR associated with ataxia with secondary vitamin E deficiency. Mov Disord 2013;28:1904-5. [PubMed: 23813796]
- Micalizzi A, Moroni I, Ginevrino M, Biagini T, Mazza T, Romani M, Valente EM. Very mild features of dysequilibrium syndrome associated with a novel VLDLR missense mutation. Neurogenetics. 2016;17:191-5. [PubMed: 27251579]
- Moheb LA, Tzschach A, Garshasbi M, Kahrizi K, Darvish H, Heshmati Y, Kordi A, Najmabadi H, Ropers HH, Kuss AW. Identification of a nonsense mutation in the very low-density lipoprotein receptor gene (VLDLR) in an Iranian family with dysequilibrium syndrome. Eur J Hum Genet. 2008;16:270-3. [PubMed: 18043714]
- Ozcelik T, Akarsu N, Uz E, Caglayan S, Gulsuner S, Onat OE, Tan M, Tan U. Mutations in the very low-density lipoprotein receptor VLDLR cause cerebellar hypoplasia and quadrupedal locomotion in humans. Proc Natl Acad Sci USA. 2008;105:4232-6. [PMC free article: PMC2393756] [PubMed: 18326629]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Schlotawa L, Hotz A, Zeschnigk C, Hartmann B, Gärtner J, Morris-Rosendahl D. Cerebellar ataxia, mental retardation and dysequilibrium syndrome 1 (CAMRQ1) caused by an unusual constellation of VLDLR mutation. J Neurol. 2013;260:1678-80. [PubMed: 23670308]
- Sonmez FM, Gleeson JG, Celep F, Kul S. The very low density lipoprotein receptor-associated pontocerebellar hypoplasia and dysmorphic features in three Turkish patients. J Child Neurol. 2013;28:379-83. [PMC free article: PMC4442636] [PubMed: 22532556]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197-207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Tissir F, Goffinet AM. Reelin and brain development. Nat Rev Neurosci. 2003;4:496-505. [PubMed: 12778121]
- Türkmen S, Guo G, Garshasbi M, Hoffmann K, Alshalah AJ, Mischung C, Kuss A, Humphrey N, Mundlos S, Robinson PN. CA8 mutations cause a novel syndrome characterized by ataxia and mild mental retardation with predisposition to quadrupedal gait. PLoS Genet. 2009;5:e1000487. [PMC free article: PMC2677160] [PubMed: 19461874]
- Türkmen S, Hoffmann K, Demirhan O, Aruoba D, Humphrey N, Mundlos S. Cerebellar hypoplasia, with quadrupedal locomotion, caused by mutations in the very low-density lipoprotein receptor gene. Eur J Hum Genet. 2008;16:1070-4. [PubMed: 18364738]
- Valence S, Garel C, Barth M, Toutain A, Paris C, Amsallem D, Barthez MA, Mayer M, Rodriguez D, Burglen L. RELN and VLDLR mutations underlie two distinguishable clinico-radiological phenotypes. Clin Genet. 2016;90:545-9. [PubMed: 27000652]
- Wilker M, Hans-Jürgen C, Schuster S, Abicht A, Boltshauser E. VLDLR-associated pontocerebellar hypoplasia with nonprogressive congenital ataxia and a diagnostic neuroimaging pattern. Neuropediatrics. 2019;50:404-5 [PubMed: 31261436]
Publication Details
Author Information and Affiliations
Children's Hospital of Eastern Ontario
Professor of Pediatrics
University of Ottawa
Ottawa, Ontario, Canada
Children's Hospital of Eastern Ontario
Ottawa, Ontario, Canada
Alberta Children's Hospital
Associate Professor of Medical Genetics
University of Calgary
Calgary, Alberta, Canada
Publication History
Initial Posting: August 26, 2008; Last Update: February 27, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Boycott KM, MacDonald SK, Parboosingh JS. VLDLR Cerebellar Hypoplasia. 2008 Aug 26 [Updated 2020 Feb 27]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
