Clinical Findings Associated with Classic CDPX2
Growth deficiency / short stature. The most common nonspecific skeletal manifestation in individuals with CDPX2 is short stature [Cañueto et al 2012]. Reported heights range from the 10th-25th percentile to 6 SD below the mean.
Craniofacial appearance. The face and head are often asymmetric. Most individuals with CDPX2 have a depressed bridge and frontal bossing. Other distinctive features include downslanting palpebral fissures, hypertelorism, low-set ears, and high-arched palate [Happle 1979, Herman 2000].
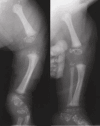
Skeletal. Stippling (chondrodysplasia punctata) most commonly involving the epiphyses of the long bones, but also the ribs, vertebrae, and tracheal cartilage is seen on radiographs in almost 100% of symptomatic infants; however, this could reflect ascertainment bias. Epiphyseal stippling can be detected on prenatal ultrasound from the second trimester [Lefebvre et al 2015] and is present in infancy and variably in childhood (during endochondral bone formation). It is usually radiologically absent in adults with CDPX2.
Approximately 90% of individuals have asymmetric (or occasionally symmetric) shortening of limbs involving mostly the femur, humerus, and other tubular bones [Happle 1979].
Moderate-to-severe kyphoscoliosis is common and can present in infancy or early childhood. Lung disease may develop secondary to progressive kyphoscoliosis and can lead to death [Sutphen et al 1995]. Spinal deformities can progress rapidly; in addition, progressive deformity following surgical vertebral fusion is common [Mason et al 2002]. Contractures, other joint abnormalities, dislocated patella and hips, and postaxial polydactyly have also been reported [Happle 1979, Has et al 2000, Herman 2000, Cañueto et al 2012, Cardoso et al 2014].
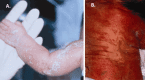
Skin, hair, and nails. The skin is involved in more than 95% of individuals of CDPX2 [Cañueto et al 2012]. Scaling ichthyosis on an erythematous base is present in newborns in a linear or blotchy pattern. The ichthyosis follows the lines of Blaschko and has a feather-like edge, but total scaling erythroderma also occurs. As the rash fades in the first weeks or months of life, it leaves a linear or whorled pattern of atrophoderma predominantly near hair follicles where scales had been located. Some individuals also have ichthyosis and/or pigmentary abnormalities that persist into childhood and adulthood.
Hair findings include scarring alopecia in patches, sparse eyelashes and eyebrows, and coarse, lusterless hair.
Minor nail findings include flattening and splitting of the nail plates [Happle 1979, Herman 2000, Hoang et al 2004].
Ocular. Approximately two thirds of individuals have cataracts at birth or develop them early in life. Cataracts are usually unilateral, asymmetric, and/or sectorial [Happle 1979, Happle 1981, Herman et al 2002]. Other eye findings include microphthalmia and/or microcornea.
Neurologic. Intelligence is typically normal in affected individuals unless a CNS malformation is present. Rarely reported neurologic abnormalities in males include posterior fossa arachnoid cysts and medullary atrophy secondary to atlas hypoplasia [Horinouchi et al 2019].
Ear anomalies and hearing. Rarely, dysplastic auricles and sensorineural hearing loss have been reported in affected individuals [Happle 1979, Herman et al 2002, Ozyurt et al 2015].
Other findings
Individuals with CDPX2 may also have bilateral or unilateral clubfoot [
Herman et al 2002].
Mortality. Typically, life expectancy is normal in individuals with CDPX2, although severe scoliosis may compromise heart and lung function and negatively affect life expectancy.