Summary
Clinical characteristics.
The findings in X-linked chondrodysplasia punctata 2 (CDPX2) range from fetal demise with multiple malformations and severe growth retardation to much milder manifestations, including females with no recognizable physical abnormalities. At least 95% of live-born individuals with CDPX2 are female. Characteristic features include growth deficiency; distinctive craniofacial appearance; chondrodysplasia punctata (stippling of the epiphyses of the long bones, vertebrae, trachea, and distal ends of the ribs); often asymmetric rhizomelic shortening of limbs; scoliosis; linear or blotchy scaling ichthyosis in the newborn; later appearance of linear or whorled atrophic patches involving hair follicles (follicular atrophoderma); coarse hair with scarring alopecia; and cataracts.
Diagnosis/testing.
The diagnosis of CDPX2 is established in a female proband with: typical clinical findings, increased concentration of 8(9)-cholestenol and 8-dehydrocholesterol in plasma, scales from skin lesions, or cultured lymphoblasts or fibroblasts; and/or a heterozygous pathogenic variant in EBP identified by molecular genetic testing.
The diagnosis of CDPX2 is established in a male proband with: typical clinical findings, increased concentration of 8(9)-cholestenol and 8-dehydrocholesterol in plasma, scales from skin lesions, or cultured lymphoblasts or fibroblasts; and/or a hemizygous pathogenic variant in EBP identified by molecular genetic testing.
Management.
Treatment of manifestations: Treatment is symptomatic and individualized. For individuals with typical CDPX2 diagnosed in the newborn period, the following are appropriate: orthopedic management of leg length discrepancy; frequent assessment of kyphoscoliosis; management of respiratory compromise as per pulmonologist; dermatologic management with emollients and keratolytics; sun protection; cataract extraction and correction of vision; standard interventions for hearing loss and hydronephrosis; family support.
Surveillance: Regular orthopedic evaluations to monitor kyphoscoliosis, joint problems, and any leg length discrepancy; follow up with a dermatologist; regular follow up of ophthalmologic abnormalities; audiology evaluations as indicated; monitor hydronephrosis if present.
Agents/circumstances to avoid: Prolonged sun exposure for individuals with ichthyosis, who are at risk of dehydration secondary to overheating. Use of emollients (which are oil based) and direct sun exposure can lead to sunburn.
Genetic counseling.
CDPX2 is inherited in an X-linked manner with early gestational male lethality. Women with an EBP germline pathogenic variant have a 50% chance of transmitting the pathogenic variant to each child: EBP pathogenic variants in sons are usually lethal; daughters will have a range of possible phenotypic expression. When the parents are clinically unaffected, the risk to the sibs of a proband appears to be low but greater than that of the general population. If the pathogenic variant cannot be detected in the DNA extracted from the leukocytes of either parent of the proband, three possible explanations are germline mosaicism, somatic mosaicism, or a de novo pathogenic variant in the proband. Prenatal diagnosis for pregnancies at increased risk is possible if the family-specific pathogenic variant is known.
Diagnosis
X-linked chondrodysplasia punctata 2 (CDPX2) is a skeletal dysplasia that also affects the skin and eyes. Specific diagnostic criteria for CDPX2 have not been published. Classic CDPX2 occurs almost exclusively in females. There are reports of affected males with an XXY karyotype [Sutphen et al 1995] or with somatic mosaicism [Aughton et al 2003, Tan et al 2010] who have clinical manifestations similar to affected females.
Suggestive Findings
CDPX2 should be suspected in an individual with the following clinical findings:
- Growth deficiency / short stature
- Craniofacial findings
- Frontal bossing
- Depressed nasal bridge
- Sparse eyebrows and lashes, often asymmetric
- Skeletal abnormality
- Stippling (chondrodysplasia punctata) involving the epiphyses of the long bones and vertebrae, the trachea, and distal ends of the ribs seen on x-ray. The presence of stippling is age dependent and cannot be seen once normal epiphyseal ossification progresses during childhood (see Figure 1).
- Rhizomelic (i.e., proximal) shortening of limbs that is often asymmetric, but occasionally symmetric
- Scoliosis, occasionally congenital
- Postaxial polydactyly (uncommon)
- Abnormality of skin, hair, and nails
- Scaling ichthyosis on an erythematous base arranged in a linear or blotchy pattern in the newborn period (following lines of Blaschko) that usually resolves in the first months of life and may be followed by linear or whorled atrophic patches involving hair follicles (follicular atrophoderma) (see Figure 2) and/or pigmentary abnormalities
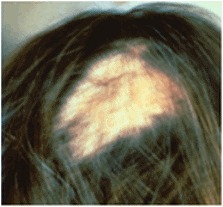
- Coarse scalp hair with scarring alopecia (see Figure 3)
- Occasional flattened or split nails
Note: Teeth are normal. - Ocular anomaly
- Cataracts often congenital, asymmetric, and/or sectorial
- Microphthalmia and/or microcornea
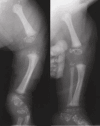
Figure 1.
Radiographs from a female infant with CDPX2 demonstrating epiphyseal stippling (also called chondrodysplasia punctata; punctate epiphyseal dysplasia) Radiograph originally published in Herman [2000]; reproduced with permission from Elsevier Ltd.
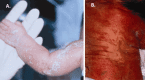
Figure 2
A. Typical skin findings of CDPX2 at birth, including scaling and an erythematous eruption that follows lines of Blaschko B. Later hyperpigmentation over the back in a two-month-old female

Figure 3.
Scarring, patchy alopecia in a female with CDPX2 Photograph originally published in Herman [2000]; reproduced with permission from Elsevier Ltd.
Establishing the Diagnosis
Female proband. The diagnosis of CDPX2 is established in a female proband with typical clinical findings, characteristic results on biochemical testing (see Table 1), and/or a heterozygous pathogenic variant in EBP identified by molecular genetic testing (see Table 2).
Male proband. The diagnosis of CDPX2 is established in a male proband (with a 46,XY karyotype) with the typical clinical findings, characteristic results on biochemical testing (see Table 1), and/or a mosaic hemizygous pathogenic variant in EBP identified by molecular genetic testing (see Table 2).
Note: Males with non-mosaic hypomorphic EBP pathogenic variants have MEND (male EBP disorder with neurologic defects) syndrome, a clinically distinct phenotype comprising neurologic and structural malformations without chondrodysplasia punctata [Arnold et al 2012].
Biochemical testing. Sterol analysis of plasma, scales from skin lesions, or cultured lymphoblasts or fibroblasts can be used for diagnosis. Increased concentrations of 8(9)-cholestenol and 8-dehydrocholesterol are essentially diagnostic of CDPX2 [Kelley et al 1999] (Table 1); however, individuals with molecularly confirmed CDPX2 with normal biochemical profiles have been reported [Whittock et al 2003]. There is no correlation between the levels of plasma 8(9)-cholestenol and 8-dehydrocholesterol and mutational subgroups or specific phenotypic traits [Has et al 2002]. In affected males, biochemical testing will not be of use in differentiating CDPX2 from MEND syndrome (see Genetically Related Disorders).
Table 1.
Concentrations of 8(9)-Cholestenol and 8-Dehydrocholesterol Observed in Chondrodysplasia Punctata 2, X-Linked
Histologic examination of skin biopsies of individuals with CDPX2 show dilated ostial hyperkeratosis of the hair follicle with keratin calcium deposits [Leclerc-Mercier et al 2015]. These findings are relatively specific for CDPX2 and have been reported in only a couple of other conditions. Skin biopsy from the affected area may therefore be a useful diagnostic adjunct in individuals with a milder presentation.
Molecular Genetic Testing
In centers where it is readily available, molecular genetic testing should be considered first. Biochemical testing can be used to support a diagnosis in individuals with inconclusive molecular variants.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of CDPX2 is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas male probands or those in whom the diagnosis of CDPX2 has not been considered due to atypical findings are more likely to be diagnosed using genomic testing (see Option 2).
Option 1. When the phenotypic and laboratory findings suggest the diagnosis of CDPX2 molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Perform sequence analysis of EBP to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected.
- A multigene panel that includes EBP and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2. When the diagnosis of CDPX2 is not considered because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is the most commonly used genomic testing method; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 2.
Molecular Genetic Testing Used in Chondrodysplasia Punctata 2, X-Linked
Clinical Characteristics
Clinical Description
Variability in females. At least 95% of individuals with X-linked chondrodysplasia punctata 2 (CDPX2) are female. The clinical phenotypes in heterozygous females are highly variable and depend on the pattern of X-chromosome inactivation in relevant tissues (i.e., percentage of active X chromosomes with the pathogenic variant) and other possible modifying factors. Phenotypes range from fetal demise with multiple malformations and severe growth retardation to much milder manifestations, such as adults with only cutaneous features, short stature, or no recognizable physical abnormalities. Severity in females varies greatly within families and among individuals with the same pathogenic variant, as would be expected for a pathogenic process determined, in part, by the random process of X-chromosome inactivation.
Phenotypes in males. Although CDPX2 was for many years presumed to be lethal in males, a small number of affected males have been reported. Sutphen et al [1995] described a male with CDPX2 and a 47,XXY karyotype. Males with mosaic EBP variants have also been reported [Aughton et al 2003, Tan et al 2010, Arnold et al 2012, Pacault et al 2018, Honigman et al 2019, Horinouchi et al 2019] with allele fractions ranging from 20% to 75% detected in blood and skin [Pacault et al 2018, Honigman et al 2019, Horinouchi et al 2019]. The clinical characteristics of males with mosaic EBP pathogenic variants are well within the marked variability described in affected females.
Clinical Findings Associated with Classic CDPX2
Growth deficiency / short stature. The most common nonspecific skeletal manifestation in individuals with CDPX2 is short stature [Cañueto et al 2012]. Reported heights range from the 10th-25th percentile to 6 SD below the mean.
Craniofacial appearance. The face and head are often asymmetric. Most individuals with CDPX2 have a depressed bridge and frontal bossing. Other distinctive features include downslanting palpebral fissures, hypertelorism, low-set ears, and high-arched palate [Happle 1979, Herman 2000].
Skeletal. Stippling (chondrodysplasia punctata) most commonly involving the epiphyses of the long bones, but also the ribs, vertebrae, and tracheal cartilage is seen on radiographs in almost 100% of symptomatic infants; however, this could reflect ascertainment bias. Epiphyseal stippling can be detected on prenatal ultrasound from the second trimester [Lefebvre et al 2015] and is present in infancy and variably in childhood (during endochondral bone formation). It is usually radiologically absent in adults with CDPX2.
Approximately 90% of individuals have asymmetric (or occasionally symmetric) shortening of limbs involving mostly the femur, humerus, and other tubular bones [Happle 1979].
Moderate-to-severe kyphoscoliosis is common and can present in infancy or early childhood. Lung disease may develop secondary to progressive kyphoscoliosis and can lead to death [Sutphen et al 1995]. Spinal deformities can progress rapidly; in addition, progressive deformity following surgical vertebral fusion is common [Mason et al 2002]. Contractures, other joint abnormalities, dislocated patella and hips, and postaxial polydactyly have also been reported [Happle 1979, Has et al 2000, Herman 2000, Cañueto et al 2012, Cardoso et al 2014].
Skin, hair, and nails. The skin is involved in more than 95% of individuals of CDPX2 [Cañueto et al 2012]. Scaling ichthyosis on an erythematous base is present in newborns in a linear or blotchy pattern. The ichthyosis follows the lines of Blaschko and has a feather-like edge, but total scaling erythroderma also occurs. As the rash fades in the first weeks or months of life, it leaves a linear or whorled pattern of atrophoderma predominantly near hair follicles where scales had been located. Some individuals also have ichthyosis and/or pigmentary abnormalities that persist into childhood and adulthood.
Hair findings include scarring alopecia in patches, sparse eyelashes and eyebrows, and coarse, lusterless hair.
Minor nail findings include flattening and splitting of the nail plates [Happle 1979, Herman 2000, Hoang et al 2004].
Ocular. Approximately two thirds of individuals have cataracts at birth or develop them early in life. Cataracts are usually unilateral, asymmetric, and/or sectorial [Happle 1979, Happle 1981, Herman et al 2002]. Other eye findings include microphthalmia and/or microcornea.
Neurologic. Intelligence is typically normal in affected individuals unless a CNS malformation is present. Rarely reported neurologic abnormalities in males include posterior fossa arachnoid cysts and medullary atrophy secondary to atlas hypoplasia [Horinouchi et al 2019].
Ear anomalies and hearing. Rarely, dysplastic auricles and sensorineural hearing loss have been reported in affected individuals [Happle 1979, Herman et al 2002, Ozyurt et al 2015].
Other findings
- Individuals with CDPX2 may also have bilateral or unilateral clubfoot [Herman et al 2002].
- Hydronephrosis has been seen in several affected females [Herman et al 2002].
- Hypoglycemia in the neonatal period has been reported [Cañueto et al 2012, Horinouchi et al 2019].
Mortality. Typically, life expectancy is normal in individuals with CDPX2, although severe scoliosis may compromise heart and lung function and negatively affect life expectancy.
Genotype-Phenotype Correlations
Null EBP variants are associated with a severe CDPX2 phenotype in females and result in intrauterine lethality in males (except in a mosaic state).
Hypomorphic hemizygous EBP variants are associated with a milder MEND phenotype in males (see Genetically Related Disorders) [Barboza-Cerda et al 2014, Barboza-Cerda et al 2019].
Penetrance
A few clinically unaffected females with molecularly confirmed CDPX2 have been reported [Herman et al 2002, Shirahama et al 2003, Hellenbroich et al 2007]. However, these females were ascertained from segregation testing, and phenotyping may not have been as comprehensive as in the proband. Some women have been so mildly affected that they were identified only after having had a child with more severe features in whom CDPX2 was diagnosed. Although these adult women have subtle findings, their findings are sufficient to consider them affected.
Nomenclature
CDPX2 has also been referred to as:
- Conradi-Hünermann syndrome, named after Conradi [1914] and Hünermann [1931], who described the first persons with this disorder;
- Happle syndrome, named after Rudolph Happle [Happle 1979, Happle 1981], who contributed greatly to characterization of the phenotype and delineation of the X-linked mode of inheritance and possible etiologies of the syndrome [Traupe 1999, Sheffield 2001];
- Conradi-Hünermann-Happle syndrome, which recognizes all three individuals who helped to define this disorder.
Prevalence
Prevalence is unknown and incidence is estimated at 1:100,000 to 1:200,000 births.
Genetically Related (Allelic) Disorders
MEND syndrome (OMIM 300960). Non-mosaic hypomorphic EBP pathogenic variants are associated with the distinct, primarily neurologic phenotype, MEND (male EBP disorder with neurologic defects) syndrome. Reported males have moderate-to-severe developmental delay and almost all have clinically important CNS malformations, most notable Dandy-Walker variant, agenesis of the corpus callosum, and major gyral abnormalities. Other unique findings include facial dysmorphisms (prominent nasal bridge, low-set ears, and large anterior fontanelle), skeletal findings (2-3 toe syndactyly, postaxial polydactyly), and urogenital findings (cryptorchidism, hypospadias) [Arnold et al 2012, de Almeida et al 2017], which clinically overlap with Smith-Lemli Opitz syndrome. Many hemizygous males have chronic ichthyosis, but, as would be predicted, not in patchy distributions. Individuals with MEND syndrome also have elevated 8(9)-cholestenol and 8-dehydrocholesterol. Epiphyseal stippling is not visible on radiographs in individuals with MEND syndrome [de Almeida et al 2017]. Females heterozygous for a MEND syndrome-related EBP pathogenic variant typically do not have clinical manifestations of the disorder.
Differential Diagnosis
Several disorders demonstrate features similar to those of X-linked chondrodysplasia punctata 2 (CDPX2) and/or manifest stippling on radiographs and various combinations of limb asymmetry, short stature, intellectual disability, cataracts, and skin changes. The key radiologic finding of chondrodysplasia punctata occurs in various metabolic disorders, skeletal dysplasias, chromosome abnormalities, and teratogenic exposures.
Genetic Disorders
Table 3.
Disorders and Genes of Interest in the Differential Diagnosis of Chondrodysplasia Punctata 2, X-Linked
Chondrodysplasia punctata, tibia-metacarpal (OMIM 118651) and humero-metacarpal types are inherited in an autosomal dominant manner. The associated genes are unknown. Affected individuals have short limbs due primarily to shortening of the tibiae/humeri, metacarpals, and phalanges. Vertebral anomalies are also found. CDP is usually confined to the sacral, carpal, and tarsal areas. No skin or eye changes are present; intellect and life expectancy is normal [Savarirayan et al 2004].
Astley-Kendall dysplasia has been postulated to be an allelic disorder of Greenberg dysplasia [Author, personal communication]. Ossification defects of the cranial vault, spine, and long bones result in shortened and dysplastic long bones and vertebrae in affected individuals.
Teratogen Exposures
Warfarin embryopathy and other vitamin K deficiencies (including vitamin K epoxide reductase deficiency) are phenotypically similar to X-linked chondrodysplasia punctata 1 with especially severe hypoplasia of the nasal bone ("Binder anomaly"), distal phalangeal abnormalities, and punctata of the axial skeleton.
Maternal autoimmune disease (systemic lupus erythematosus [SLE], mixed connective tissue disease and scleroderma) can cause CDP with rhizomelic limb shortening.
Management
No published guidelines exist to establish the extent of disease or proper management in an individual with X-linked chondrodysplasia punctata 2 (CDPX2). The following recommendations are based on current literature and the authors' experience.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with CDPX2, the evaluations summarized in Table 4 (if they have not already been completed) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with Chondrodysplasia Punctata 2, X-Linked
Treatment of Manifestations
Treatment is symptomatic and should be tailored to the individual.
Table 5.
Treatment of Manifestations in Individuals with Chondrodysplasia Punctata 2, X-Linked
Surveillance
Table 6.
Recommended Surveillance for Individuals with Chondrodysplasia Punctata 2, X-Linked
Agents/Circumstances to Avoid
Adequate sun protection is recommended for individuals with ichthyosis, who are at risk of dehydration secondary to overheating during prolonged sun exposure. Furthermore, care must be taken with use of emollients (which are oil based) and direct sun exposure, which can lead to sunburn.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
X-linked chondrodysplasia punctata 2 (CDPX2) is inherited in an X-linked manner with typical, but not absolute, male lethality.
Risk to Family Members
Parents of a female proband
- A female proband may have inherited the EBP pathogenic variant from either her mother or her father, or the pathogenic variant may be de novo.
- The mother of a proband may be so mildly affected that she is identified only after having had a child with more severe features in whom CDPX2 was diagnosed.
- If the proband’s father is asymptomatic, it is possible (though not likely) that he has the pathogenic variant in some cells of his body (somatic and germline mosaicism).
- Detailed evaluation of the parents and review of the extended family history may help distinguish probands with a de novo pathogenic variant from those with an inherited pathogenic variant.
- Molecular genetic testing of the mother (and possibly the father, or subsequently the father) can help to determine if the pathogenic variant was inherited. If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, possible explanations include a de novo pathogenic variant in the proband or germline mosaicism in a parent. Parental somatic and/or germline mosaicism has been described in rare families [Has et al 2002, Morice-Picard et al 2011, Pacault et al 2018].
Parents of a male proband
- Because almost all males with classic CDPX2 are mosaic for a pathogenic variant in EBP (presumably the result of a de novo postzygotic change), the mother of a male with classic CDPX2 will typically not be heterozygous for the pathogenic variant.
- In rare families, the mother of an affected male may be heterozygous or may have germline mosaicism. To date, only one family has been reported in which a male had nonlethal CDPX2 as the result of a constitutional EBP pathogenic variant inherited from his heterozygous mother [Bode et al 2013].
- In a family with more than one affected individual, the mother of an affected male is an obligate heterozygote. Note: If a woman has more than one affected child and no other affected relatives and if the EBP pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism.
- The father of an affected male will not have the disorder nor will he be hemizygous for the EBP pathogenic variant; therefore, he does not require further evaluation/testing.
Sibs of a proband
- The risk to sibs of a female proband depends on the genetic status of the parents.
- The risk to sibs of a male proband is presumed to be low, as the majority of males with CDPX2 have the disorder as the result of a postzygotic mosaic pathogenic variant.
- If the mother of the proband has an EBP pathogenic variant, the chance of transmitting it in each pregnancy is 50%.
- Males who inherit the pathogenic variant will be affected. CDPX2 is associated with early gestational male lethality, although a small number of males with constitutional CDPX2-causing pathogenic variants have been reported.
- Females who inherit the pathogenic variant will be heterozygotes and will have a range of clinical manifestations (see Clinical Description).
- If the father of the proband has an EBP pathogenic variant, he will transmit it to all his daughters and none of his sons.
- If the proband represents a simplex case (i.e., a single occurrence in a family) and if the EBP pathogenic variant cannot be detected in the leukocyte DNA of either parent, the risk to sibs is slightly greater than that of the general population because of the possibility of parental germline mosaicism.
Offspring of a proband
- Women with a CDPX2-causing pathogenic variant have a 50% chance of transmitting the pathogenic variant to each child.
- Males who inherit the pathogenic variant will be affected. CDPX2 is associated with early gestational male lethality*; males with inherited non-mosaic hypomorphic pathogenic variants who survive have MEND syndrome (see Genetically Related Disorders).* Males with mosaic EBP variants have been reported (see Clinical Description, Phenotypes in males); mosaic pathogenic variants in affected males are not inherited and are presumed to be secondary to a postzygotic mutation.
- Females who inherit the pathogenic variant will be heterozygotes and will have a range of clinical manifestations (see Clinical Description).
- Affected males transmit the pathogenic variant to all of their daughters and none of their sons. To the authors' knowledge, no affected males with constitutional CDPX2-causing pathogenic variants have been reported to reproduce. Some reported males with mosaic EBP variants have transmitted the pathogenic variant to their daughters [Has et al 2002, Pacault et al 2018].
Other family members. If the mother (or, in rare cases, the father) of the proband has an EBP pathogenic variant, her (or his) family members may be at risk of being affected.
Note: Molecular genetic testing may be able to identify the family member in whom a de novo pathogenic variant arose, information that could help determine genetic risk status of the extended family.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, have the pathogenic variant, or are at risk of having the pathogenic variant.
Prenatal Testing and Preimplantation Genetic Testing
Once the EBP pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for CDPX2 are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- FIRST - Foundation for Ichthyosis and Related Skin TypesPhone: 215-997-9400; 800-545-3286Email: info@firstskinfoundation.org
- Little People of AmericaPhone: 888-LPA-2001; 714-368-3689Fax: 707-721-1896Email: info@lpaonline.org
- MAGIC FoundationPhone: 800-362-4423Email: contactus@magicfoundation.org
- UCLA International Skeletal Dysplasia Registry (ISDR)Phone: 310-825-8998
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Chondrodysplasia Punctata 2, X-Linked: Genes and Databases
Table B.
OMIM Entries for Chondrodysplasia Punctata 2, X-Linked (View All in OMIM)
Molecular Pathogenesis
X-linked chondrodysplasia punctata 2 (CDPX2) is caused by a deficiency of 3-beta-hydroxysteroid-delta(8),delta(7)-isomerase or "sterol-Δ8-isomerase," which is encoded by EBP [Braverman et al 1999, Derry et al 1999]. Sterol-Δ8-isomerase is thought to be an integral endoplasmic reticulum membrane protein that converts 8(9)-cholestenol to lathosterol during cholesterol biosynthesis.
Variability in phenotypes among females with CDPX2 is likely attributable to variability in X-chromosome inactivation.
Mechanism of disease causation. Loss of function.
Chapter Notes
Author History
Melissa A Dempsey, MS, CGC; University of Chicago (2011-2020)
Gail E Herman, MD, PhD; Ohio State University (2011-2020)
Smitha Kumble, MBBS (2020-present)
Ravi Savarirayan, MBBS, MD, FRACP, ARCPA (Hon) (2020-present)
Christopher Tan, MS, CGC; University of Chicago (2011-2020)
Revision History
- 9 January 2020 (sw) Comprehensive update posted live
- 31 May 2011 (me) Review posted live
- 13 July 2009 (md) Original submission
References
Literature Cited
- Arnold AW, Bruckner-Tuderman L, Has C, Happle R. Conradi-Hunermann-Happle syndrome in males vs. MEND syndrome (male EBP disorder with neurological defects). Br J Dermatol. 2012;166:1309–13. [PubMed: 22229330]
- Aughton DJ, Kelley RI, Metzenberg A, Pureza V, Pauli RM. X-linked dominant chondrodysplasia punctata (CDPX2) caused by single gene mosaicism in a male. Am J Med Genet A. 2003;116A:255–60. [PubMed: 12503102]
- Barboza-Cerda MC, Barboza-Quintana O, Martínez-Aldape G, Garza-Guajardo R, Déctor MA. Phenotypic severity in a family with MEND syndrome is directly associated with the accumulation of potentially functional variants of cholesterol homeostasis genes. Mol Genet Genomic Med. 2019;7:e931. [PMC free article: PMC6732292] [PubMed: 31397093]
- Barboza-Cerda MC, Wong LJ, Martínez-de-Villarreal LE, Zhang VW, Déctor MA. A novel EBP c.224T>A mutation supports the existence of a male-specific disorder independent of CDPX2. Am J Med Genet A. 2014;164A:1642–7. [PubMed: 24700572]
- Bode H, Galm C, Hummler H, Teller C, Haas D, Gencik M. Non-lethal non-mosaic male with Conradi-Hunermann syndrome caused by a novel EBP c.356T>G mutation. Am J Med Genet A. 2013;161A:2385–8. [PubMed: 23852825]
- Braverman N, Lin P, Moebius FF, Obie C, Moser A, Glossmann H, Wilcox WR, Rimoin DL, Smith M, Kratz L, Kelley RI, Valle D. Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hünermann syndrome. Nat Genet. 1999;22:291–4. [PubMed: 10391219]
- Cañueto J, Girós M, Ciria S, Pi-Castán G, Artigas M, García-Dorado J, García-Patos V, Virós A, Vendrell T, Torrelo A, Hernández-Martín A, Martín-Hernández E, Garcia-Silva MT, Fernández-Burriel M, Rosell J, Tejedor M, Martínez F, Valero J, García JL, Sánchez-Tapia EM, Unamuno P, González-Sarmiento R. Clinical, molecular and biochemical characterization of nine Spanish families with Conradi-Hünermann-Happle syndrome: new insights into X-linked dominant chondrodysplasia punctata with a comprehensive review of the literature. Br J Dermatol. 2012;166:830–8. [PubMed: 22121851]
- Cardoso ML, Barbosa M, Serra D, Martins E, Fortuna A, Reis-Lima M, Bandeira A, Balreira A, Marques F. Living with inborn errors of cholesterol biosynthesis: lessons from adult patients. Clin Genet. 2014;85:184–8. [PubMed: 23509885]
- Conradi E. Vorzeitiges aufreten von knochen und eigenartigen verkalkungskernen bei chondrodystrophia fotalis hypoplastica histologische und rontgenuntersuchungen. Jb Kinderheilk. 1914;80:86–97.
- de Almeida H Jr, Has C, Rampon G, Isaacsson H, de Castro LA. MEND syndrome: a case report with scanning electron microscopy findings of the collodion membrane. Acta Derm Venereol. 2017;97:110–1. [PubMed: 27276700]
- Derry JM, Gormally E, Means GD, Zhao W, Meindl A, Kelley RI, Boyd Y, Herman GE. Mutations in a delta 8-delta 7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. Nat Genet. 1999;22:286–90. [PubMed: 10391218]
- Happle R. Cataracts as a marker of genetic heterogeneity in chondrodysplasia punctata. Clin Genet. 1981;19:64–6. [PubMed: 7460383]
- Happle R. X-linked dominant chondrodysplasia punctata: review of literature and report of a case. Hum Genet. 1979;53:65–73. [PubMed: 535904]
- Has C, Bruckner-Tuderman L, Muller D, Floeth M, Folkers E, Donnai D, Traupe H. The Conradi-Hunerman-Happle syndrome (CDPX2) and emopamil binding protein: novel mutations, and somatic and gonadal mosaicism. Hum Mol Genet. 2000;9:1951–5. [PubMed: 10942423]
- Has C, Seedorf U, Kannenberg F, Bruckner-Tuderman L, Folkers E, Fölster-Holst R, Baric I, Traupe H. Gas chromatography-mass spectrometry and molecular genetic studies in families with the Conradi-Hunermann-Happle syndrome. J Invest Dermatol. 2002;118:851–8. [PubMed: 11982764]
- Hellenbroich Y, Grzeschik KH, Krapp M, Jarutat T, Lehrmann-Petersen C, Buiting K, Gillessen-Kaesbach G. Reduced penetrance in a family with X-linked dominant chondrodysplasia punctata. Eur J Med Genet. 2007;50:392–8. [PubMed: 17625999]
- Herman GE. X-linked dominant disorders of cholesterol biosynthesis in man and mouse. Biochim Biophys Acta. 2000;1529:357–73. [PubMed: 11111102]
- Herman GE, Kelley RI, Pureza V, Smith D, Kopacz K, Pitt J, Sutphen R, Sheffield LJ, Metzenberg AB. Characterization of mutations in 22 females with X-linked dominant chondrodysplasia punctata (Happle syndrome). Genet Med. 2002;4:434–8. [PubMed: 12509714]
- Hoang MP, Carder KR, Pandya AG, Bennett MJ. Ichthyosis and keratotic follicular plugs containing dystrophic calcification in newborns: Distinctive histopathologic features of X-linked dominant chondrodysplasia punctata (Conradi-Hunermann-Happle syndrome). Am J Dermatopathol. 2004;26:53–8. [PubMed: 14726822]
- Honigman A, De Cruz R, Sinclair R, Winship I. Chondrodysplasia punctata (CDPX2) in a male caused by single-gene mosaicism: A 20-year follow-up. Australas J Dermatol. 2019;60:e160–e162. [PubMed: 30294782]
- Horinouchi T, Morisada N, Uemura H, Kobayashi D, Nozu K, Okamoto N, Iijima K. Male CDPX2 patient with EBP mosaicism and asymmetrically lateralized skin lesions with strict midline demarcation. Am J Med Genet A. 2019;179:1315–8. [PubMed: 31034146]
- Hünermann C. Chondrodysplasia calcificans congeneita als abortive form der chondrodystrophia. Z Kindergeilkd. 1931;51:1–19.
- Kelley RI, Wilcox WG, Smith M, Kratz LE, Moser A, Rimoin DS. Abnormal sterol metabolism in patients with Conradi-Hunerman-Happle syndrome and sporadic lethal chondrodysplasia punctata. Am J Med Genet. 1999;83:213–19. [PubMed: 10096601]
- König A, Happle R, Bornholdt D, Engel H, Grzeschik KH. Mutations in the NSDHL gene, encoding a 3beta-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am J Med Genet. 2000;90:339–46. [PubMed: 10710235]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–5. [PMC free article: PMC3965032] [PubMed: 24234437]
- Leclerc-Mercier S, Dufernez F, Fraitag S, Coulombe J, Dompmartin A, Barreau M, Bozon D, Lamazière A, Bonnefont JP, Khalifa E, Bodemer C, Hadj-Rabia S. Keratotic follicular plugs with calcifications in Conradi-Hunermann-Happle syndrome: histological, biochemical and genetic testing correlation. Br J Dermatol. 2015;173:1316–8. [PubMed: 26075358]
- Lefebvre M, Dufernez F, Bruel AL, Gonzales M, Aral B, Saint-Onge J, Gigot N, Desir J, Daelemans C, Jossic F, Schmitt S, Mangione R, Pelluard F, Vincent-Delorme C, Labaune JM, Bigi N, D'Olne D, Delezoide AL, Toutain A, Blesson S, Cormier-Daire V, Thevenon J, El Chehadeh S, Masurel-Paulet A, Joyé N, Vibert-Guigue C, Rigonnot L, Rousseau T, Vabres P, Hervé P, Lamazière A, Rivière JB, Faivre L, Laurent N, Thauvin-Robinet C. Severe X-linked chondrodysplasia punctata in nine new female fetuses. Prenat Diagn. 2015;35:675–84. [PubMed: 25754886]
- Lykissas MG, Sturm PF, McClung A, Sucato DJ, Riordan M, Hammerberg KW. Challenges of spine surgery in patients with chondrodysplasia punctata. J Pediatr Orthop. 2013;33:685–93. [PubMed: 23836071]
- Mason DE, Sanders JO, MacKenzie WG, Nakata Y, Winter R. Spinal deformity in chondrodysplasia punctata. Spine. 2002;27:1995–2002. [PubMed: 12634559]
- Morice-Picard F, Kostrzewa E, Wolf C, Benlian P, Taïeb A, Lacombe D. Evidence of postzygotic mosaicism in a transmitted form of Conradi-Hunermann-Happle syndrome associated with a novel EBP mutation. Arch Dermatol. 2011;147:1073–6. [PubMed: 21931045]
- Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, Nishimura G, Robertson S, Sangiorgi L, Savarirayan R, Sillence D, Superti-Furga A, Unger S, Warman ML. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019;179:2393–419. [PubMed: 31633310]
- Ozyurt K, Subasioglu A, Ozturk P, Inci R, Ozkan F, Bueno E, Cañueto J, González Sarmiento R. Emopamil binding protein mutation in Conradi-Hunermann-Happle syndrome representing plaque-type psoriasis. Indian J Dermatol. 2015;60:216. [PMC free article: PMC4372958] [PubMed: 25814754]
- Pacault M, Vincent M, Besnard T, Kannengiesser C, Bénéteau C, Barbarot S, Latypova X, Belabbas K, Lamazière A, Winer N, Joubert M, Bézieau S, Isidor B, Mercier S, Nizon M, Leclerc-Mercier S, Hadj-Rabia S, Dufernez F. New splicing pathogenic variant in EBP causing extreme familial variability of Conradi-Hunermann-Happle Syndrome. Eur J Hum Genet. 2018;26:1784–90. [PMC free article: PMC6244079] [PubMed: 30135486]
- Savarirayan R, Boyle RJ, Masel J, Rogers JG, Sheffield LJ. Longterm follow-up in chondrodysplasia punctata, tibia-metacarpal type, demonstrating natural history. Am J Med Genet A. 2004;124A:148–57. [PubMed: 14699613]
- Sheffield LJ. Comment on Traupe’s tribute to Rudolf Happle. Am J Med Genet. 2001;101:283. [PubMed: 11424147]
- Shirahama S, Miyahara A, Kitoh H, Honda A, Kawase A, Yamada K, Mabuchi A, Kura H, Yokoyama Y, Tsutsumi M, Ikeda T, Tanaka N, Nishimura G, Ohashi H, Ikegawa S. Skewed X-chromosome inactivation causes intra-familial phenotypic variation of an EBP mutation in a family with X-linked dominant chondrodysplasia punctata. Hum Genet. 2003;112:78–83. [PubMed: 12483303]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197-207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Sutphen R, Amar MJ, Kousseff BG, Toomey KE. XXY male with X-linked dominant chondrodysplasia punctata (Happle syndrome). Am J Med Genet. 1995;57:489–92. [PubMed: 7677157]
- Tan C, Haverfield E, Dempsey M, Kratz L, Descartes M, Powell B. X-linked dominant chondrodysplasia punctata and EBP mutations in males. Poster session. Albuquerque, NM: American College of Medical Genetics Annual Clinical Genetics Meeting; 2010.
- Traupe H. Functional X-chromosomal mosaicism of the skin: Rudolph Happle and the lines of Blaschko. Am J Med Genet. 1999;85:324–9. [PubMed: 10398252]
- Whittock NV, Izatt L, Mann A, Homfray T, Bennett C, Mansour S, Hurst J, Fryer A, Saggar AK, Barwell JG, Ellard S, Clayton PT. Novel mutations in X-linked dominant chondrodysplasia punctata (CDPX2). J Invest Dermatol. 2003;121:939–42. [PubMed: 14632217]
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: May 31, 2011; Last Update: January 9, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Kumble S, Savarirayan R. Chondrodysplasia Punctata 2, X-Linked. 2011 May 31 [Updated 2020 Jan 9]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.