Polysaccharide Capsules
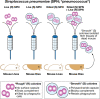
A human in good health is colonized by as many as 1013 bacteria on the skin and mucosal surfaces, particularly in the gastrointestinal tract, a number that resembles the number of cells in our own body. Despite all these direct and continuous encounters, it is relatively rare that bacteria invade into the tissues to produce serious infections. Although not restricted to pathogenic species/strains, one feature shared by many of these disease-causing agents is the presence of diverse polysaccharide capsule structures, which encapsulate bacteria (Chapter 21). These structures can have many biological properties, both stimulating and thwarting immune detection in different settings. Capsules make key contributions to the virulence of multiple bacterial pathogens, reducing recognition by the innate and adaptive immune systems. However, capsular polysaccharides are also commonly the natural targets of the adaptive immune system. In fact, antibodies specific to different capsule structures often define the serology used to differentiate subtypes of particular bacterial species. Streptococcus pneumoniae (pneumococcus) encodes perhaps the most historically significant capsule in biology and medicine. Pioneering experiments by Frederick Griffith in 1928 showed the in vivo transfer of virulence from a dead encapsulated (smooth) disease-causing strain to an avirulent, nonencapsulated (rough) strain. Studies regarding immunogenicity of pneumococcal polysaccharides (Figure ) provided the framework for the discoveries of Oswald Avery, Colin MacLeod, and Maclyn McCarty, which showed DNA to be the carrier of genetic information.
Classical experiments on the role of the pneumococcal polysaccharide capsule in virulence. Streptococcus pneumoniae (SPN) strains can be identified with either a “rough” (R) or a “smooth” (S) phenotype, the latter being (more...)
Killing of bacteria by phagocytes of the innate immune system, such as neutrophils or macrophages, is aided by opsonization, a process in which the bacterial surface is tagged with complement proteins or specific antibodies. Phagocytes express receptors for activated complement and antibody Fc domains, allowing them to bind, engulf, and kill bacteria. Together these processes are referred to as opsonophagocytosis. Genetic mutagenesis of capsule biosynthesis genes and infectious challenge in small animal models have illustrated the roles of bacterial capsules in resisting opsonophagocytosis. Compared with the wild-type parent bacterial strains, isogenic capsule-deficient mutants of group A Streptococcus (GAS), group B Streptococcus (GBS), pneumococcus, Haemophilus influenzae, Neisseria meningitidis (meningococcus), Salmonella serotype Typhi (typhoid fever), Bacillus anthracis (anthrax), and several other important human pathogens are rapidly cleared from the bloodstream by opsonophagocytosis and fail to establish systemic infection. Some bacteria mimic the anionic host molecule sialic acid in their capsules, including the neonatal pathogens GBS and Escherichia coli K1. These sialylated capsules bind the complement regulatory protein factor H and attenuate complement deposition. Independent of the complement system, GBS sialic acids also reduce neutrophil bactericidal activities by directly engaging the sialic acid–binding receptor Siglec-9.
Bacteria also use molecular mimicry to evade antibody generation by the adaptive immune system. Generally, humans can generate effective antibodies against bacterial polysaccharide capsules, but this ability is diminished early and late in life. Infants and the elderly are particularly prone to invasive infections with encapsulated pathogens. Molecular mimicry of common host glycan structures allows bacteria to masquerade as “self” to avoid being recognized by the adaptive immune system. For example, the GAS pathogen expresses a nonimmunogenic capsule of hyaluronan, identical to the nonsulfated glycosaminoglycan that is highly abundant in host skin and cartilage (Chapter 16). The contribution of capsule-based host mimicry to bacterial immune evasion is also illustrated by the homopolymeric sialic acid capsules of E. coli and meningococcus, an important cause of sepsis and meningitis. Whereas the group C meningococcal capsule is composed of an α2-9-linked sialic acid polymer that is a unique bacterial structure, the group B meningococcal capsule is composed of an α2-8-linked sialic acid polymer that is identical to a motif present on neural cell adhesion molecules (NCAMs) found in human neural tissues (Chapter 15). The group C capsule has proven to be a successful vaccine antigen in human populations, whereas the group B capsule is essentially nonimmunogenic. Bacterial polysaccharide capsules can also cloak immunogenic surface proteins to which antibodies might be directed.
One of the challenges posed to the host is that different strains of the same bacterial species can display diverse compositions and linkages of repeating sugar units in their capsule structures. Often, these structures are immunologically distinct, allowing classification of different capsule “serotype” strains. For example, there are five major capsule serotypes of meningococcus (A, B, C, Y, and W-135), six different capsule serotypes of the respiratory pathogen H. influenzae (a–f), nine capsule serotypes of GBS (Ia, Ib, and II–VIII), and more than 90 different serotypes of pneumococcus, which is a leading cause of bacterial pneumonia, sepsis, and meningitis. Antibodies generated by the host against the capsule of one serotype strain typically do not provide cross-protective immunity. Thus, individuals can be repeatedly infected over their lifetime by different serotypes of the same bacterial pathogen. Figuratively, although the strategy of capsular molecular mimicry used for example by GAS renders the pathogen invisible to immune surveillance, the strategy of antigenic diversity of capsule types presents a moving target to the immune system. Genetic exchange of capsule biosynthetic genes among serotype strains of an individual species (e.g., the polysialyltransferase gene of meningococcus) can lead to capsule switching in vivo, which provides another means of pathogen escape from protective immunity.
Lipopolysaccharide
In addition to a polysaccharide capsule, Gram-negative bacteria have an outer membrane that is rich in lipopolysaccharide (LPS; Chapter 21), also known as endotoxin. As its name suggests, LPS consists of two parts, a lipid A moiety and glycan components. Lipid A is comprised of two glucosamines, acyl chains and phosphates, which are embedded in the outer membrane. The glycan components extend outward into the bacterial niche. A core oligosaccharide contains some sugars not found in vertebrates (such as ketodeoxyoctulonate [Kdo] and heptose) and a repeating polysaccharide known as the O-antigen that can vary widely among strains within an individual species. Many mucosal pathogens such as H. influenzae, Campylobacter jejuni, and Neisseria gonorrhoeae lack O-antigens; instead, they produce lipooligosaccharides (LOSs) that contain only lipid A and an extended core structure.
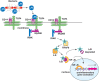
LPS is a pathogen-associated molecular pattern (PAMP) that is recognized by the innate immune system and stimulates inflammatory processes, including the classic fever response. Bacteria that have breached the barrier defenses of the skin or mucosa release soluble LPS, which is recognized by membrane receptors CD14 and Toll-like receptor 4 (TLR4), initiating downstream immune signaling processes (Figure ). TLR4 belongs to an evolutionarily conserved family of receptors (TLRs) that can detect a wide array of microbe-derived ligands. For example, the TLR2 receptor can recognize peptidoglycan or lipoteichoic acid derived from the cell walls of Gram-positive bacteria that generally lack LPS. A signaling cascade ultimately leads to the activation of the transcription factor nuclear factor-κB (NF-κB). Translocation of NF-κB into the nucleus leads to the regulation of cellular processes ranging from immune cell differentiation, activation of the inflammasome, and up-regulation of proinflammatory chemokines and cytokines, including tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1).
Activation of immune signaling by bacterial lipopolysaccharide (LPS). LPS from the cell wall of Gram-negative bacteria is bound by the pattern-recognition molecule Toll-like receptor 4 (TLR4) in conjunction with the cell-surface receptor CD14. The binding (more...)
Although TLR-mediated detection of LPS and other microbial molecules is critical for triggering host innate immunity, a dangerous condition known as sepsis can develop in the setting of overwhelming bacterial infections that lead to dysregulated systemic immune responses. Symptoms include fever, low blood pressure, rapid heart rate, abnormal white blood cell counts, and dysfunction of multiple organ systems that may lead to lung or kidney failure and death.
Processes of selection have led to many examples of Gram-negative bacteria that vary or modify the structure of LPS to interfere with antibiotic action and host defenses. For example, some LPS modifications reduce the overall negative charge of LPS (e.g., as phosphoethanolamine or 4-amino-4-deoxy-L-arabinose [L-Ara4N]) and in doing so can repel cationic antimicrobial peptides of host and microbial origin, such as host defensins or bacterial polymyxins. The plague bacillus Yersinia pestis changes the number and type of acyl groups on the lipid A of its LPS in response to temperature changes. At environmental temperatures (∼21°C), Y. pestis, which lacks O-antigens, predominantly expresses a more immunostimulatory hexa-acylated lipid A, whereas at mammalian body temperature (37°C), the pathogen expresses a less immunostimulatory tetra-acylated lipid A. These data suggest that the production of a less immunostimulatory form of LPS on entry into a mammalian host might confer avoidance to immune detection.
Sialic acids and related nonulosonic acids are also synthesized de novo or scavenged from the host for incorporation into the LOS of several Gram-negative pathogens, including pathogenic members of the genera Haemophilus, Neisseria, Campylobacter, and Vibrio. This can be accomplished by several distinct mechanisms and often confers properties of immune evasive and enhanced virulence. For example, the human-specific pathogen N. gonorrhoeae uses a surface sialyltransferase and scavenges the activated form of sialic acid (CMP-Neu5Ac) from the host. Such incorporation thwarts multiple arms of the host complement system and confers resistance to the cationic antimicrobial peptides (CAMPS). An Achilles’ heel of this increasingly drug-resistant pathogen may be the promiscuity of its sialyltransferase toward related nonulosonic acids. Unnatural incorporation of legionaminic acid (CMP-Leg5,7Ac2) in N. gonorrhoeae prevents the immune evasion benefits typically afforded by Neu5Ac; moreover, vaginal administration of CMP-Leg5,7Ac2 leads to more rapid bacterial clearance of genital infections in mice.
Studies of another Gram-negative bacterium, Vibrio vulnificus, underscore that the presence of legionaminic acid in LOS may not always be a deterrence to virulence. V. vulnificus is normally present in environmental (aquatic) ecosystems and is pathogenic in humans only during accidental contact with susceptible individuals, often following consumption of raw or undercooked seafood. Although it is a rare human pathogen, the bacterium can cause bloodstream and disseminated infections that are commonly fatal when they strike. Legionaminic acid contributes to the ability of V. vulnificus to survive in bloodstream and cause disseminated infection following intravenous administration.
Occasionally, the host mounts an (auto)immune response to “self” antigens, including sialylated LOS. For example, the foodborne pathogen, C. jejuni, is capable of expressing a variety of sialylated LOS structures that mimic human gangliosides (Chapters 11 and 46). Sialylation of C. jejuni LOS results in CD14-dependent amplification of mucosal dendritic cell (DC) responses, which promote B-cell proliferation in a T-cell-independent manner. These events may help explain why in a small subset of infections, self-reacting (autoimmune) antibodies are inappropriately produced by B cells, leading to the attack of nerve fibers expressing the relevant gangliosides. These events can result in a life-threatening paralytic disorder known as Guillain–Barré syndrome (GBS). For most patients, GBS symptoms have to be managed by removing cross-reactive antibodies (plasmapheresis) or immunomodulatory therapies such as intravenous immunoglobulin (IVIg).