

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
The NCBI Handbook [Internet]. 2nd edition. Bethesda (MD): National Center for Biotechnology Information (US); 2013-.
This publication is provided for historical reference only and the information may be out of date.

The BLAST Sequence Analysis Tool
Thomas Madden, PhD.
Author Information and Affiliations 1.
1.Created: March 15, 2013.
Estimated reading time: 14 minutes
Scope
A sequence similarity search often provides the first information about a new DNA or protein sequence. A search allows scientists to infer the function of a sequence from similar sequences. There are many ways of performing a sequence similarity search, but probably the most popular method is the “Basic Local Alignment Search Tool” (BLAST) (1, 2). BLAST uses heuristics to produce results quickly. It also calculates an “expect value” that estimates how many matches would have occurred at a given score by chance, which can aid a user in judging how much confidence to have in an alignment.
As the name implies, BLAST performs “local” alignments. Most proteins are modular in nature, with one or more functional domains occurring within a protein. The same domains may also occur in proteins from different species. The BLAST algorithm is tuned to find these domains or shorter stretches of sequence similarity. The local alignment approach also means that an mRNA can be aligned with a piece of genomic DNA, as is frequently required in genome assembly and analysis. If instead BLAST started out by attempting to align two sequences over their entire lengths (known as a global alignment), fewer similarities would be detected, especially with respect to domains and motifs.
There are many different flavors of BLAST searches:
- The BLASTN nucleotide-nucleotide search looks for more distant sequences.
- BLASTP performs protein-protein sequence comparison, and its algorithm is the basis of many other types of BLAST searches such as BLASTX and TBLASTN.
- BLASTX searches a nucleotide query against a protein database, translating the query on the fly.
- TBLASTN searches a protein query against a nucleotide database, translating the database on the fly.
- PSI-BLAST first performs a BLASTP search to collect information that it then uses to produce a Position-Specific-Scoring-Matrix (PSSM). A PSSM for a query of length N is an N x 20 matrix. Each of the N columns corresponds to a letter in the query, and each column contains 20 rows. Each row corresponds to a specific residue and describes the probability of related sequences having that residue at that position. PSI-BLAST can then search a database of protein sequences with this PSSM.
- RPSBLAST (Reverse-Position-Specific BLAST) can very quickly search a protein query against a database of PSSMs that were usually produced by PSI-BLAST.
- DELTA-BLAST produces a PSSM with a fast RPSBLAST search of the query, followed by searching this PSSM against a database of protein sequences (5).
A brief summary of “how BLAST works” will assist the reader in understanding the rest of this chapter. BLAST uses heuristics, which really means that it takes shortcuts to get to the proper answer faster. It is useful to break the BLAST search down into a few different phases called setup, preliminary search, and traceback. In the setup phase, BLAST reads in the query, search parameters, and database. It may first check the query for low-complexity or other repeats, and then produces a set of “words”--short, fixed-length sequences based on the query. They are used to initiate matches in the database (or “subject”) sequences. In the preliminary search, a number of steps are performed on every sequence in the database. First, the database is scanned for matches to the words, and those are used to initiate a gap-free extension. Second, gap-free extensions that achieve a certain score are used to seed a gapped extension that only calculates the score and extent and leaves to a later stage the time- and memory-consuming work of calculating insertions and deletions. Gapped extensions that achieve a specified score are saved, though lower-scoring matches may be deleted if too many matches are found. In the final traceback phase of the search, gapped extensions saved in the preliminary phase are used as seeds for a gapped extension that also calculates the insertions and deletions and may use more sensitive parameters. More details on the BLAST algorithm are provided in (1, 2).
BLAST provides a variety of ways to perform a search:
- NCBI BLAST website at http://blast.ncbi.nlm.nih.gov (6-8). The website requires no setup or registration, is simple to use, produces results quickly, and requires only a web browser. The BLAST website is a shared public resource, so users who send in many hundreds or thousands of searches in a short time may run afoul of usage guidelines.
- BLAST URL API. The URL API documentation describes the URL parameters of the website that will not change and can be used in the future. This API also uses a shared public resource, so users should respect usage guidelines.
- BLAST standalone applications. Users with specialized or proprietary data, or who require resources beyond what NCBI can provide, can use the BLAST+ applications to run BLAST in “standalone mode,” (9) on their own computers. BLAST+ in standalone mode uses data on local servers, using the user’s own data and/or using BLAST databases downloaded from NCBI. To use standalone mode, users must have sufficient computational resources for their searches. BLAST databases can be large (many gigabytes), and setting up a standalone BLAST+ environment requires some effort. On the other hand, standalone BLAST+ may be the best option for sophisticated users. BLAST+ applications run on Mac OSX, Windows, and most flavors of LINUX/UNIX. Instructions on the use of BLAST+ can be found at https://www.ncbi.nlm.nih.gov/books/NBK279690/
- BLAST+ remote service. Users who need to do many searches at the NCBI, or who want to script searches, can use the remote service available with the BLAST+ applications. Instructions on the use of the remote service with the BLAST+ applications can be found at https://www.ncbi.nlm.nih.gov/books/NBK279690/ The remote service also uses a shared public resource, so users should respect usage guidelines.
- C++ Application Programming Interface (API). For very specialized applications, the NCBI C++ Toolkit offers a programmatic interface to BLAST described at https://ncbi.github.io/cxx-toolkit/pages/ch_blast The API supports both standalone and remote searches (at the NCBI).
This chapter focuses on the specifics of how BLAST works, most especially at the NCBI, and how to avoid using it incorrectly. There are links to documentation and videos about using BLAST at http://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastDocs.
History
NCBI produced the first version of BLAST around 1990, accompanied by the publication of the first BLAST paper by Altschul et al. (2). This version performed only gap-free alignments, but provided p-values that allowed users to judge the statistical significance of their result. A gapped version of BLAST appeared in 1997 (1). This release also included PSI-BLAST, which could produce a PSSM and then search the database with it. Both of these BLAST versions made use of the NCBI C toolkit. In late 2009, the NCBI started supporting a newer version of BLAST (called BLAST+) (9) based upon the NCBI C++ toolkit as a development platform. Afterwards, the C toolkit and the older BLAST packages were deprecated and users were strongly encouraged to use the BLAST+ applications for standalone needs. The NCBI BLAST website was built with the C++ toolkit and BLAST+.
Data Model
Structured Output: Flexible Results
Most users who are familiar with the BLAST report think of it as the output from BLAST, but the real picture is somewhat more complicated. A BLAST search first produces results as structured output, which permits automatic and rigorous checks for syntax errors and changes. Typical report formats such as the BLAST or GenBank report do not permit such automatic checks. BLAST output can be represented as XML or ASN.1 (Abstract Syntax Notation 1), enabling automated syntax and structure validation. For example, the structure of an XML document can be ensured by validating it against its DTD or Schema. ASN.1, used extensively at the NCBI since 1990, is also constrained by a module definition (similar to a schema), and its binary format is very compact, making it ideal for transmission over networks.
BLAST search results are first represented as C++ objects, which can be used directly to output data formatted for further processing. Since many of the objects have ASN.1 representations, they can also be serialized to ASN.1 and written to disk or sent over a network. These serialized objects can then be used to recreate the original C++ objects at a different time or place.
There are a few advantages to this procedure. First, results that are serialized to disk can be formatted later. Second, results that are sent over a network can be formatted on another machine. Third, serialized results can be formatted multiple times in different ways using the same ASN.1 objects. Finally, since many NCBI tools produce objects corresponding to the ASN.1 modules, they have a straightforward way to exchange results.
While ASN.1 makes storage and transmission of BLAST results efficient, the XML representations of these objects provides a bridge to common XML tool chains. The choice of which to use depends on the application.
The Alignment: Data You Really Need
BLAST outputs its alignments using the ASN.1 SeqAlign module. A SeqAlign indicates where an alignment starts and ends on sequences, provides the coordinates of insertions and deletions, and (for DNA) tells the strand to which the query sequence aligned. It also lists scores, expect values, and sequence identifiers for the alignment. A SeqAlign does not contain the actual query or database sequences, or other information such as the titles for any sequences, but only the sequence identifiers. In addition to the alignment data, BLAST reports require other information about the database sequence, such as the actual sequence, title, and taxonomy information. Since those data don’t occur in the SeqAlign, BLAST report formatters retrieve sequence data as needed, using the sequence identifier, from the BLAST database or from Entrez.
The Rest: Data You Might Need
In addition to alignment data, BLAST also produces other outputs that characterize the search inputs and results:
The Blast4-request ASN.1 type contains the query, the search parameters, and the database. It is often referred to as a “search strategy”. Both the website and the standalone applications can produce the search strategy as an ASN.1 object, and the website can store it as a “Saved search”. The user may use the search strategy to repeat the search at a later time, with optional changes to the query or database.
The Blast4-archive ASN.1 type contains the query and search information as a search strategy, the alignment information as a SeqAlign, the location of any masks applied to the query (for low-complexity or interspersed repeats), and the Karlin-Altschul parameters used to calculate statistics.
The search strategy and Blast4-archive types depend on the ASN.1 module defined for the BLAST+ remote service, which is the network interface of the NCBI BLAST queuing system Splitd (described below).
Dataflow
Searches at the NCBI: How It’s Done
The NCBI uses a custom queuing system called Splitd to schedule searches that were submitted via the website or BLAST+ remote service (Figure 1). The Splitd system first parses the input from a user and produces an ASN.1 object with the relevant information that is stored in an MSSQL database. Some searches require a preparatory step such as retrieval of data from Entrez. In that case, a daemon requests the data from Entrez after verifying that it has not been cached by a recent search. Another example with a preparatory step is the RPSBLAST search used to produce a PSSM by DELTA-BLAST.
Once any preparatory steps have completed, the Splitd server queues the search. Queuing priority depends partly on whether the user has other searches queued: the search may be penalized (i.e., put back further in the queue) if the user has many uncompleted searches. Splitd spreads each search over multiple machines (each of which is running a backend), where each backend searches only a part of the database. The partial result produced by a backend is called a chunk. The backend performs only the setup and preliminary search (as described above), so its final product is a set of matches listing scores, extents, and database sequences identifiers for those matches. Results from the backends are forwarded to a merger, which collects all chunks in memory, merges them into a single result, and writes the complete set of preliminary results to an MSSQL database. These results are then sent to the traceback daemon, which performs the final step of producing the alignment that includes insertions and deletions, final scores, and extents. Finally, the traceback is written to the MSSQL database and the search is marked as finished. The user can then format the search in a variety of formats as shown in Figure 2.
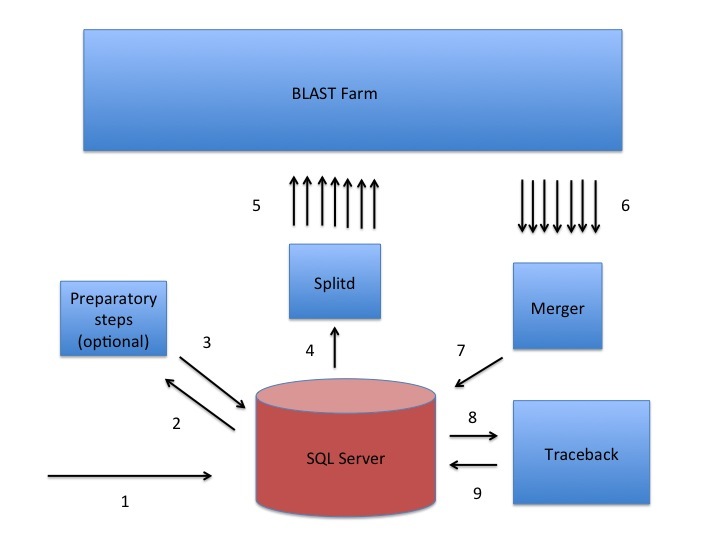
Figure 1:
A sketch of the Splitd system used at the NCBI for processing BLAST requests. The numbers in the figure identify steps in the process. First, the search is inserted into the SQL database as ASN.1 [1]. Second, an optional preparatory step may retrieve data from Entrez or produce a PSSM using RPSBLAST [2, 3]. Third, Splitd pulls the search from the SQL database [4], queues it, and spreads it across several backends [5]. The search is processed on the BLAST farm. Results from backends are preliminary, because they include only the extent of alignments as well as the scores, but no insertions or deletions. Next, these results are sent to the Merger [6]. The Merger merges all pieces into one result which it stores in the SQL server [7]. The traceback then pulls the preliminary results from the SQL database and produces results that include insertions and deletions [8]. Finally, the traceback places the full results into the SQL database [9]. At this point the user may retrieve the results using the Request ID that the system issued.
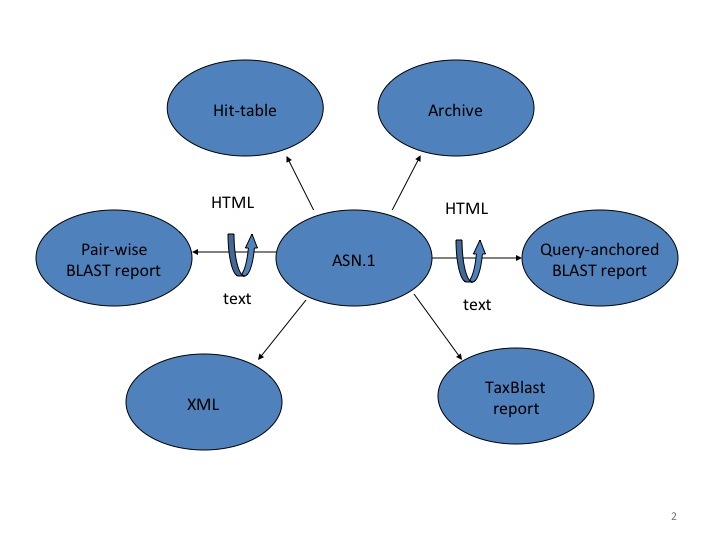
Figure 2:
Different output formats that can be generated from the Splitd ASN.1. Most reports require additional information not stored in the ASN.1, such as database sequences or taxonomy information retrieved from other sources.
BLAST Databases: Some Details to Keep in Mind
Generally, BLAST does not directly search GenBank flatfiles. Rather, sequences are transformed into BLAST databases with a special format that makes searching more efficient. The BLAST indexing processes splits and indexes the sequence records, producing several files. The “header” and the “sequence” files are the most important ones. The header file contains information such as the sequence title and taxonomy information and is used mostly during formatting of the BLAST report. The sequence file contains the sequence information and is used most heavily during the BLAST search. DNA has a small alphabet (four letters, if there are no ambiguities) so the DNA sequence file consumes a little more than one byte per four bases. A BLAST database is normally partitioned into multiple volumes, with each volume representing a contiguous subset of the database. The size of the volume can be specified when the database is created, but the NCBI has found that a volume size of about one gigabyte works well. For the Sequence Read Archive (SRA), BLAST searches the underlying SRA objects directly. This is efficient because the SRA objects group the data in a manner similar to that of BLAST.
As mentioned above, the results produced by BLAST (e.g., the SeqAlign) do not contain the database sequences, but only identifiers for them. BLAST must use the sequence identifiers to retrieve the sequence data from some other source, such as the BLAST database or Entrez. This means that an identifier must uniquely identify a sequence in the database. Furthermore, the query sequence should not have the same identifier as any sequence in the database, unless the query sequence itself is in the database. Any BLAST database or FASTA file from the NCBI website that contains GI numbers already satisfies the uniqueness criterion. Ambiguous identifiers are normally a problem only when custom databases are produced and care is not taken in assigning identifiers. The identifier for a FASTA entry is the first token (meaning the letters up to the first space) after the > sign on the definition line. The simplest case is to simply provide a unique token (e.g., 1, 2, and so on), but it is possible to construct more complicated identifiers that might, for example, describe the data source. NCBI supports a specific syntax for such parsable identifiers described at https://ncbi.github.io/cxx-toolkit/pages/ch_demo#ch_demo.T5.
The makeblastdb application produces BLAST databases from FASTA files, and its –parse_seqids flag instructs makeblastdb to expect unique parsable identifiers. If the identifiers are not parsed, then makeblastdb adds some ad hoc (internal) identifiers to the BLAST database, but this may limit the options for display or further processing of the alignments in the result. The BLAST+ user manual at https://www.ncbi.nlm.nih.gov/books/NBK279690/ presents detailed instructions about how to use makeblastdb.
Many users are familiar with the process of producing the BLAST database from FASTA files, but this is not the process used to create most BLAST databases available at the NCBI. Rather, most BLAST databases on NCBI servers are produced directly from the central NCBI ID system (described in the chapter on Dataflow). Nucleotide databases at the NCBI can contain tens of billions of bases, so de novo indexing can consume significant resources and time. Therefore, most BLAST databases are updated incrementally, rather than being completely rewritten every time sequence data changes. The ID system also adds other available information (such as taxonomy) to the BLAST database. The NCBI makes many of these databases available as FASTA in the BLAST portion of the FTP site. These FASTA files are produced from the original BLAST databases.
BLAST Reports: Look Before You Parse
BLAST can produce a number of different reports, but it is important to understand the purpose of each report to use it effectively. The standard BLAST report consists of several sections, including a table of descriptions (sequence titles) and alignments. It is meant as a human-readable report, and is subject to change with little or no notice. The NCBI strongly discourages parsing this report, and provides other formats that are better suited for automated processes. One of the simplest formats is the tabular output, which provides basic information in an easy-to-parse form. As stated earlier, structured output allows for automatic and rigorous checks for syntax errors and changes. The standard BLAST report and the tabular report are not structured output, so they do not permit automated checks for syntax errors and changes. Only a structured report such as XML or ASN.1 can allow for such automated checks. The NCBI makes available a special BLAST XML report that contains much of the same data (e.g., the sequences) as the standard BLAST report, but it allows formal checks for correctness. BLAST can also produce ASN.1 output.
Access
The NCBI supports a number of methods to submit BLAST searches to the Splitd system.
The most heavily used way to submit searches is via the NCBI website at blast.ncbi.nlm.nih.gov. Figure 3 presents the submission page for nucleotide-nucleotide searches. The user simply inputs a sequence identifier or raw sequence and clicks the BLAST button. If desired, they may change the database or limit their search taxonomically using autocomplete menus (Figure 4). There are a number of other ways to modify the search, but most users make minimal changes to the defaults. At this point, the Blast.cgi script parses the input, constructs an ASN.1 representation of the search, inserts the search into an MSSQL database, and returns a Request ID (RID) to the user. The Splitd system then processes the request is as shown in Figure 1. Meanwhile, the user’s browser periodically polls the server, checking for complete results. Once the results are complete, the page displays the report, which the user may reformat as desired. Results are usually saved for 36 hours on the server. The user may use the RID to retrieve the results.
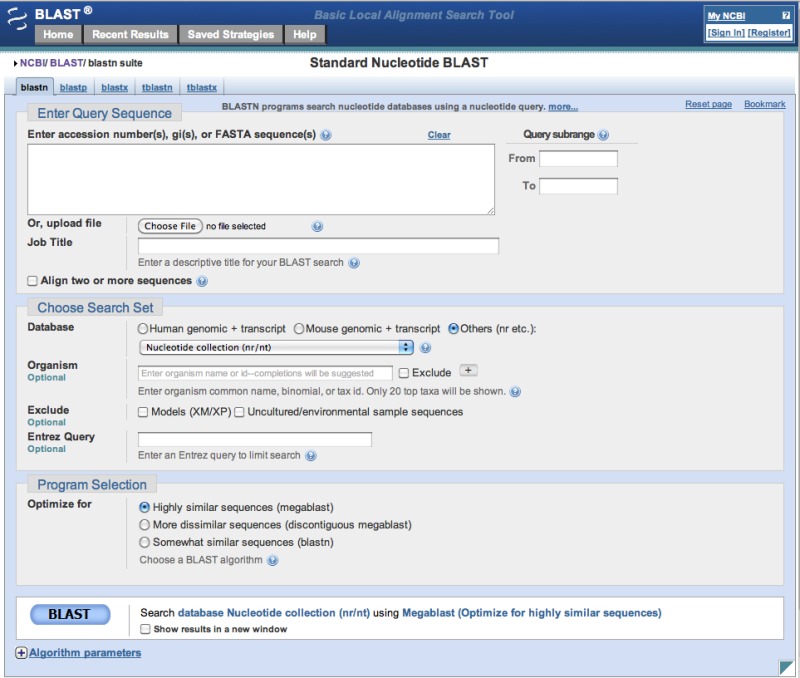
Figure 3:
Nucleotide-nucleotide search page.
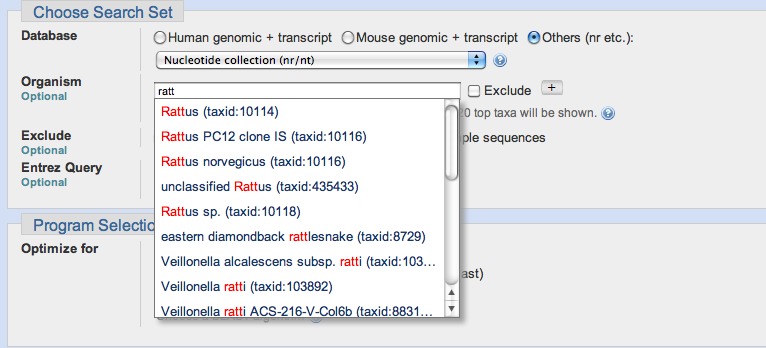
Figure 4:
Detail from the nucleotide-nucleotide search page. The autocomplete menu presents suggestions as the user types. Once a user has selected an entry, an Entrez query is constructed and processed as a “preparatory” step by the Splitd system (see Figure 1 and text).
Users may also access the NCBI BLAST service through the BLAST+ remote service, which is a network service that uses ASN.1 to communicate between the client and the server. In this case, the client sends the query, parameters, and database to the server in the form of an ASN.1 request. Splitd processes the request in the manner described previously. An RID is assigned and sent back to the client. The client polls for the status of the result on a regular basis. Once the search is done, the ASN.1 results returned to the client include the alignment (as a SeqAlign) and masking information. Because the SeqAlign does not contain the database sequences, the BLAST+ remote client fetches sequence data from the NCBI as needed for formatting.
Programmers can use the NCBI “URL API” interface and the HTTP protocol to create BLAST jobs and retrieve BLAST results. The URL API documentation describes the URL parameters of Blast.cgi that will not change and can be used in the future, and explains how to interpret BLAST results programmatically.
Related Tools
There are many tools that run BLAST searches and post-process the output for specific purposes. Three tools supported by the NCBI are:
- Primer-BLAST (10). Primer-BLAST finds primers that would amplify only a specific gene. It first uses Primer3 to identify primers on a gene sequence template, and then uses BLAST to search the template against the specified databases. It extensively post-processes the BLAST output to identify primers that uniquely amplify the desired gene. Primer-blast uses the BLAST+ remote service to send the search to Splitd and to receive results. It makes full use of the ASN.1-encoded objects to post-process BLAST results and create presentations.
- IgBLAST (11). IgBLAST annotates the variable regions of an immunoglobulin sequence, which includes a variable (V), diversity (D), and a joining (J) segment. These different segments have different characteristic lengths and require different BLAST parameters. IgBLAST orchestrates the multiple BLAST searches needed, and then presents a unified report to the user. It calls either the BLAST API directly or uses the BLAST+ remote service.
- VecScreen. The VecScreen service identifies vector contamination in a query. It uses a specialized database and extensively post-processes the initial results produced by BLAST. Information about VecScreen is available at http://www.ncbi.nlm.nih.gov/tools/vecscreen/
References
- 1.
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. [PMC free article: PMC146917] [PubMed: 9254694]
- 2.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. [PubMed: 2231712]
- 3.
- Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7(1-2):203–14. [PubMed: 10890397]
- 4.
- Morgulis A, Coulouris G, Raytselis Y, Madden TL, Agarwala R, Schaffer AA. Database indexing for production MegaBLAST searches. Bioinformatics. 2008;24(16):1757–64. [PMC free article: PMC2696921] [PubMed: 18567917]
- 5.
- Boratyn GM, Schaffer AA, Agarwala R, Altschul SF, Lipman DJ, Madden TL. Domain enhanced lookup time accelerated BLAST. Biol Direct. 2012;7:12. [PMC free article: PMC3438057] [PubMed: 22510480]
- 6.
- Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36(Web Server issue):W5-9. [PMC free article: PMC2447716] [PubMed: 18440982]
- 7.
- McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32(Web Server issue):W20-5. [PMC free article: PMC441573] [PubMed: 15215342]
- 8.
- Ye J, McGinnis S, Madden TL. BLAST: improvements for better sequence analysis. Nucleic Acids Res. 2006;34(Web Server issue):W6-9. [PMC free article: PMC1538791] [PubMed: 16845079]
- 9.
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. [PMC free article: PMC2803857] [PubMed: 20003500]
- 10.
- Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. [PMC free article: PMC3412702] [PubMed: 22708584]
- 11.
- Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41(Web Server issue):W34-40. [PMC free article: PMC3692102] [PubMed: 23671333]
- The BLAST Sequence Analysis Tool - The NCBI HandbookThe BLAST Sequence Analysis Tool - The NCBI Handbook
Your browsing activity is empty.
Activity recording is turned off.
See more...