

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
The NCBI Handbook [Internet]. 2nd edition. Bethesda (MD): National Center for Biotechnology Information (US); 2013-.
This publication is provided for historical reference only and the information may be out of date.

Scope
Sequence variation is of scientific interest to population geneticists, genetic mappers, and those investigating relationships among variation and phenotype. These variations can be of several types, from simple substitutions that do not affect sequence length, to those that result in minor length differences, to those that affect multiple genes and multiple chromosomes. Variations can also be categorized with respect to their frequency within a population, from a variation with a single allele to a variation that is highly polymorphic.
Although SNP is the abbreviation for “single nucleotide polymorphism,” dbSNP is a public archive of all short sequence variation, not just single nucleotide substitutions that occur frequently enough in a population to be termed polymorphic. dbSNP includes a broad collection of simple genetic variations such as single-base nucleotide substitutions, small-scale multi-base deletions or insertions, and microsatellite repeats. Data submitted to dbSNP can be from any organism, from any part of a genome, and can include genotype and allele frequency data if those data are available. dbSNP accepts submissions for all classes of simple sequence variation, and provides access to variations of germline or somatic origin that are clinically significant.
In order to emphasize the comprehensive nature of dbSNP’s content, the full name of the database was changed from “database of Single Nucleotide Polymorphism” to the more inclusive “database of Short Genetic Variation” in July of 2011. The acronym that represents the database will remain “dbSNP” to avoid any confusion that might arise from a complete name change.
Each record in dbSNP includes the sequence context of the variant, the frequency of the polymorphism in a population if available, its zygosity if available, and the experimental method(s), protocols, and conditions used to assay the variation by each submitter. Individual submissions are clustered into dbSNP reference records (rs#) that contain summary data which may include clinical significance from ClinVar, association with phenotype from dbGaP, variation false positive status, allele origin (germline or somatic), and submitter attributes.
The dbSNP has been designed to support submissions and research into a broad range of biological problems that include the identification of genotype-phenotype relationships, genetic and physical mapping, functional analysis, pharmacogenomics, and association studies.
Medical Genetics
Advances in next-generation sequencing technologies allow researchers to generate massive amounts of sequence data. When clinical samples are sequenced using these technologies, novel variants that have causative roles in disease may be identified. dbSNP’s role is to manage information on the location and type of these novel variants, while ClinVar manages the current interpretation of the variants' clinical phenotype.
dbSNP integrates clinical attribute data from ClinVar (i.e., clinical assertions and asserted allele origin) into new and existing human refSNP records, as well as into additional curated attribute data that includes minor allele frequency and variation false positive status.
VCF files generated from dbSNP’s curated records can be used to filter (subtract) known variants from a set of variant calls to identify novel variants or narrow a list of potential causative variants that might warrant further evaluation.
Genome Mapping
Variations are used as positional markers in genetic and physical mapping of nucleotide sequences when they map to a unique location in a genome. In other words, the variations represented by dbSNP records can serve as stable landmarks in the genome even if the variation is fixed for one allele in a sample. When multiple alleles are observed in a sample pedigree, pedigree members can be tested for variation genotypes as in traditional genetic mapping studies. To aid in such mapping efforts, dbSNP updates variation mapping and annotation for each organism with the release of every new genome assembly.
Molecular and Functional Consequences
Variations that occur in functional regions of genes or in conserved non-coding regions might affect transcription, post-transcriptional processing, or a protein product. dbSNP computes the molecular consequence of any sequence change, based on NCBI’s annotation of the genome. Functional consequences may be reported from submitters.
Association Studies
dbSNP annotates variations with significant association to phenotype from Genome Wide Association Studies (GWAS) as reported by dbGAP and provides a detailed catalog of common variations. dbSNP’s GWAS annotations and common variation catalog are used to inform the design of GWAS studies, the creation of variation arrays used in GWAS studies, and the interpretation of GWAS study results.
History
Creation and Growth
dbSNP was established in September,1998, to address the need for a general catalog of genomic variation that would facilitate the scientific community’s efforts in genetic association studies, gene mapping, and evolutionary biology. Initially, dbSNP was composed of small-scale locus specific submissions defined by flanking invariant sequence. Following the advent of high-throughput sequencing and the availability of complete genome assemblies for many organisms, however, dbSNP now receives a greater number of variants defined by sequence change at asserted locations on a reference sequence.
Evolution in Submitted Content
Because dbSNP was developed before a human reference assembly was available, initial submissions were primarily from human and defined a variant sequence in the context of flanking sequence. There was often little supporting evidence or validation data. As sequencing and other discovery technologies have changed, dbSNP has grown apace, and now includes data from over 300 organisms as well as ample validation data, including multiple independent submissions, frequency data, genotype data, and allele observations. To meet community needs for a centralized variation database, in the spring of 2008, dbSNP began accepting clinical assertions for new and existing variations as well as asserted locations for variation placement. The ClinVar database, now has the role of accepting variation clinical assertion data, and following its own accession process, will route novel variation positions to dbSNP for the assignment of ss (submitted SNP) and rs (refSNP) numbers.
In addition to integrating clinical assertions into refSNP records, dbSNP has introduced other curated attributes into refSNP records, such as minor allele frequency, asserted allele origin, and potential false positive status. It also uses the curated records to generate VCF files that can be employed to filter variation calls for the presence of novel variations and identify potential causative variants.
Usage Evolution
Originally, the data in dbSNP was used only to populate sequence maps since polymorphic marker density was too low to allow further application of the data. By 2007, however, marker density had increased enough to allow for the use of variation data in association studies, high resolution mapping, and a host of other applications, including population evolution and phylogeny studies that continue to further our understanding of genetic relationships and the genomic basis of traits.
dbSNP’s current integration of clinical information into dbSNP records will allow greater application of dbSNP data to the fields of molecular medicine and pharmacogenetics, as well as emerging fields study such as pharmacometabolomics and precision medicine.
Data Model
The dbSNP data model will evolve to capture new content, but currently has two major classes of data. The first class is submitted data, namely original observations of sequence variation, which are accessioned using a “submitted SNP” (ss) identifier. The second class is generated during the dbSNP build cycle (Figure 1) by aggregating data from multiple submissions as well as data from other sources and is identified with a “reference SNP” (rs) number. The rs identifier represents aggregation by type of sequence change and location on the genome if an assembled genome is available, or aggregation by common sequence if a genome is not available.
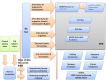
It is important to note that no matter how the data are aggregated, the rs identifier is an identifier for a location and type of variation—it is not an identifier for every sequence that may have been observed at that location. In other words, if there is a single nucleotide variation in which alleles A, C, G, and T have all been observed, they all have the same rs identifier. And, if at a location, there is a single nucleotide variation and a length variation, then multiple rs identifiers will be assigned, one for each variation type.
Note: dbSNP is in the process of updating its assembly process. Further information about assembly changes will be available on dynamic SNP documentation currently under construction.
Submitted Content
dbSNP accepts submissions from public laboratories and private organizations. dbSNP does not accept synthetic mutations or variations ascertained from cross-species alignments and analysis. Variations > 50 nucleotides in length should be submitted to the Database of Genomic Structural Variation (dbVAR).
dbSNP will not hold data to be released on a particular date or in a particular dbSNP build. If, however, you are submitting non-clinical human data or non-human data and your manuscript requires dbSNP accession numbers (ss numbers) for the review process, we can hold the submitted data until the publication is accepted and you have notified us that dbSNP can release the data. Once notification has been given, dbSNP will release the data during the next build release cycle. See the ClinVar Submission documentation for the asserted clinical variation data hold policy.
A short tag or abbreviation called a “submitter HANDLE” uniquely defines each submitting laboratory and groups the submissions within the database. See dbSNP’s online submission instructions for help preparing a submission.
The 10 major data elements of a submission include:
Sequence Context
An essential component of a submission to dbSNP is an unambiguous definition of sequence context of the variation being submitted. dbSNP no longer accepts sequence context as a variant sequence within a flanking sequence, and now minimally requires that sequence context be submitted as an asserted position on RefSeq or INSDC sequences.
Asserted Positions
Asserted positions are statements based on experimental evidence that a variant is located at a particular position on a sequence accessioned in a public database. dbSNP prefers that all variant asserted positions are submitted on a sequence accession that is part of an assembly housed in the NCBI Assembly Resource. If no assembly is available, dbSNP will accept data on a RefSeq or INSDC sequence for an asserted position that is not associated with an assembly.
For those variations that have asserted positions not associated with an assembly, the rs of that variation cannot be annotated to the assembly, and therefore will not appear on maps or graphic representations of the assembly. If, however, at some future date, a new assembly is created in the Assembly Resource to which the sequence aligns, the reported variant will be assigned an rs number at that time. Once an rs number is assigned to the variant, the variant will appear on maps or graphic representations of the assembly.
Flanking Sequence
dbSNP no longer accepts flanking sequence on a routine basis, and now requires that variant positions are reported as asserted positions on a sequence that is part of an assembly housed in the NCBI Assembly Resource.
Flanking sequence can only be used to report sequence context for those variants whose location could not be placed using an asserted position. Variations submitted with flanking sequence will be assigned a submitted SNP (ss) number that can be accessed by using the dbSNP homepage “ID search” tool or through an FTP download.
Because variants submitted with flanking sequence will be assigned an ss ID only, they will not appear on maps or graphic representations of the assembly. If, however, an assembly becomes available at a later date that allows us to map by BLAST, we will assign an rs to the variant if it is possible. dbSNP cannot predict when such an assembly will become available, or when mapping by BLAST will occur.
If a variant must be submitted with flanking sequence, dbSNP accepts variation flanking sequence as either genomic DNA or cDNA, and has a minimum length requirement of 25 bp on either side of the variation to maximize the specificity of the sequence in larger contexts.
Note: dbSNP structures its submissions so that a user can distinguish regions of assayed sequence actually surveyed for variation from those regions that are cut and pasted from a published reference sequence to satisfy dbSNP’s minimum-length requirements.
Note: SS numbers can be used in publications describing Assay Variants.
Alleles
Alleles define each variation class (Table 1). dbSNP defines single nucleotide variants in its submission scheme as G, A, T, or C, and does not permit ambiguous IUPAC codes, such as N, in the allele definition of a variation. In cases where variants occur close to one another, dbSNP permits IUPAC codes such as N, and in the flanking sequence of a variation, actually encourages them. See (Table 1) for the rules that guide dbSNP post-submission processing in assigning allele classes to each variation.
Table 1.
Variation class organizes submissions by allele definition Note: dbSNP has an allele length limitation of <=50bp. Submit alleles >50 nucleotides in length to the Database of Genomic Structural Variation (dbVAR).
Method
Each submitter defines the methods in their submission as either the techniques used to assay variation or the techniques used to estimate allele frequencies. dbSNP groups methods by method class (Table 2) to facilitate queries using general experimental technique as a query field. The submitter provides all other details of the techniques in a free-text description of the method. Submitters can also use the METHOD_EXCEPTION field to describe changes to a general protocol for particular sets of data (batch-specific details). Submitters generally define methods only once in a submission.
Table 2.
Method class organizes submissions by methodological or experimental approach
Asserted Allele Origin
A submitter can provide a statement (assertion) with supporting experimental evidence that a variant has a particular allelic origin. Assertions for a single refSNP are summarized and given an attribute value of germline or unknown. Variants of somatic origin should be submitted to ClinVar. Additional attributes (e.g., paternal) will be added in the future.
Population
Each submitter defines population samples either as the group used to initially identify variations or as the group used to identify population-specific measures of allele frequencies. These populations may be one and the same in some experimental designs. Although dbSNP has assigned populations to a population class based on the geographic origin of the sample, we will phase out this practice in the near future since most population descriptions are now submitted to BioSample. We are encouraging dbSNP submitters to start registering their samples with BioSample to obtain an assigned accession that they can use in their dbSNP submission.
Sample Size
There are two sample-size fields in dbSNP. One field, SNPASSAY SAMPLE SIZE, reports the number of chromosomes in the sample used to initially ascertain or discover the variation. The other sample size field, SNPPOPUSE SAMPLE SIZE, reports the number of chromosomes used as the denominator in computing estimates of allele frequencies. These two measures need not be the same.
Population-specific Allele Frequencies
Alleles typically exist at different frequencies in different populations; a very common allele in one population may be quite rare in another population. Also, allelic variants can emerge as private polymorphisms when particular populations have been reproductively isolated from neighboring groups, as is the case with isolated or remote populations.
Frequency data are submitted to dbSNP as allele counts or binned frequency intervals, depending on the precision of the experimental method used to make the measurement. dbSNP contains records of allele frequencies for specific population samples that are defined by each submitter and used in validating submitted variations. See Table 3 for use of allele frequencies in validation.
Table 3.
Validation status codes summarize available validation data
Population-specific Genotype Frequencies
Similar to alleles, genotypes have frequencies in populations that can be submitted to dbSNP, and are used in validating submitted variations.
Individual Genotypes
dbSNP accepts individual genotypes from samples provided by donors that have consented to having their DNA sequence housed in a public database (e.g., HapMap or the 1000 Genomes project). Genotypes reported in dbSNP contain links to population and method descriptions. General genotype data provide the foundation for individual haplotype definitions and are useful for selecting positive and negative control reagents in new experiments.
Validation Information
dbSNP accepts individual assay records (ss numbers) without validation evidence. When possible, however, dbSNP tries to distinguish high-quality validated data from unconfirmed (usually computational) variation reports. Assays validated directly by the submitter through the VALIDATION section show the type of evidence used to confirm the variation. Additionally, dbSNP will flag an assay variation as validated (Table 3) if:
- There are multiple independent submissions to the refSNP cluster with at least one non-computational method,
OR
- The variation was genotyped by the HapMap project, sequenced by the 1000 Genomes project, or other large sequencing projects.
Computed Content
dbSNP releases its content to the public in periodic “builds” that are synchronized with the release of new genome assemblies for each organism (Handbook: Eukaryotic Genome Annotation Pipeline). The dbSNP build process proceeds as follows:
- 1.
Cluster variations (ss) submitted since the previous build into RefSNPs (rs).
- 2.
Map the refSNP clusters to the appropriate assembly.
- 3.
Merge co-locating refSNP clusters when appropriate.
- 4.
Mark suspected false positive variants (see the “Suspect Variations” section of this chapter for more information on false positive selection).
- 5.
Compute a functional context for the mapped variants.
- 6.
Compute minor allele frequency as well as average heterozygosity and standard error.
- 7.
Compute links to other relevant NCBI resources such as Gene, PubMed, and RefSeq for RefSNP clusters.
- 8.
Map all clustered variations to RefSeq sequences.
See Figure 1 for a complete graphic description of the dbSNP Build process.
Dataflow
New Submissions and the Start of a New Build
Each build starts with a “close of data” that defines the set of new submissions that will be mapped to genome sequence for subsequent annotation and grouping of variations into refSNPs. The set of new data entering each build typically includes all submissions received since the close of data in the previous build.
Submitted SNPs and Reference SNP Clusters
When a new variation is submitted to dbSNP, it is assigned a unique submitted SNP ID number (ss#). If the variation is submitted with an asserted position, once the ss number is assigned, the asserted position coordinates are remapped to the corresponding coordinates on the current assembly. If the variation is submitted with flanking sequence, dbSNP aligns the flanking sequence of each submitted SNP to its appropriate genomic location(s).
When multiple submissions of the same variation class (Table 1) that have the same weight (uniqueness) map to the same position on the assembly, dbSNP clusters the ss, defines the “reference SNP cluster,” or “refSNP,” and provides the cluster with a unique RefSNP ID number (rs#). If submitted SNPs of more than one variation class map to a single position, then an rs number will be assigned for each variation class at that position. If only one submission maps to a specific position, then its ss is assigned an rs number and is the only member of its RefSNP cluster until another submitted SNP of the same variation class is found that maps to the same position.
A refSNP cluster has a number of summary attributes that are computed over all cluster members (Figure 2), and are used to annotate variations contained in other NCBI resources. See Figures 2A, 2B, 2C and 2D for the location of all summary attributes and internal/external resource links in a refSNP cluster report.
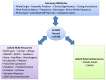

Figure 2A.
The refSNP Summary Report: Allele Summary and Integrated Maps Sections. The Allele Summary section of the refSNP report provides clinical significance (A), where the phenotype may be viewed by clicking on either the VarView or the OMIM buttons; the allele (more...)
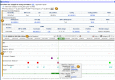
Figure 2B.
The refSNP Summary Report: the GeneView Section. The GenView section has a Display menu at the top. Once menu choices have been made, clicking the “Go” button (A) will generate the GeneView Display. The default setting (“Clinical (more...)

Figure 2C.
The refSNP Summary Report: The GeneView Display. The default GeneView display provides a tabular summary of variations mapped to splice variants of a particular gene. The variation summary is arranged in the sequential order that the variations appear (more...)

Figure 2D.
The refSNP Summary Report: FASTA, Resource Links, Population Diversity and Validation Summary Sections. The FASTA section provides the variation 5’ flanking sequence (A), the allele (B), and the 3’ flanking sequence (C) as provided by (more...)
dbSNP exports the entire dbSNP refSNP set in many report formats to its FTP site, and delivers them as sets of results when a user conducts a dbSNP batch query.
Note: Summary properties derived from asserted data (e.g., clinical assertions, asserted positions, asserted allele origin) are based on experimental evidence and cannot be seen as a confirmation of a particular clinical phenotype, genomic position, or allele origin since NCBI does not independently verify assertions and cannot endorse their accuracy.
Mapping to a Genome Sequence
When a new genome build is ready, dbSNP uses assembly-assembly alignments to remap ss asserted locations and rs locations from the old assembly to the new assembly.
To map submissions without asserted locations to a genome assembly, dbSNP obtains FASTA files for those submitted SNPs submitted prior to the “close of data,” as well as FASTA files for refSNPs in the current build that can’t be remapped, and then maps the submitted SNPs and refSNPs to the genome sequence using the BLAST procedure described in Appendix 2.
If an organism is represented by multiple assemblies then each assembly is annotated. For example, dbSNP annotates two major human assemblies: the Genome Reference Consortium (GRC) Reference assembly, and the haploid hydatidiform mole (CHM1) assembly.
refSNP Clustering and refSNP Orientation
The orientation of a refSNP, and hence its sequence and allele string, is set by the first submitted SNP (ss) used to create a refSNP (rs) cluster. By convention, the cluster exemplar is the member of a refSNP cluster that has the longest flanking sequence or is the first variant with an asserted location assigned in the cluster. If in a later build, a new variant added to the cluster becomes the exemplar and happens to be in reverse orientation to the current orientation of the refSNP, dbSNP preserves the orientation of the refSNP by using the reverse complement of the cluster exemplar to set the orientation of the refSNP sequence.
For those variants that are submitted with an asserted position rather than a flanking sequence, once dbSNP maps and verifies the asserted position, the flanking sequence is derived from the asserted position and used to determine the variant orientation.
Once the clustering process determines the orientation of all member sequences in a cluster, it will gather a comprehensive set of alleles for a refSNP cluster.
Following the dbSNP redesign, dbSNP will keep the use of flanking sequence and exemplars for those organisms that don’t have a mature assembly, but will phase out the “exemplar” concept once creation of asserted positions for those variations that were submitted with flanking sequence has begun.
Note: dbSNP reports all variants and variant alleles on the + strand of the assembly.
Re-Mapping, refSNP Merging and refSNP Splitting
Re-Mapping and refSNP Merging
RefSNPs are operationally defined as a variation at a location on a reference assembly. Every time there is a genomic assembly update, the interim reference sequence may change, so the refSNPs must be updated or re-clustered.
The re-clustering process begins when NCBI updates the genomic assembly. All existing refSNPs (rs) and newly submitted SNPs (ss) are mapped to the genome assembly using assembly-assembly remap or multiple BLAST and MegaBLAST cycles as delineated in Appendix 2.
dbSNP clusters variations that co-locate at the same place on the genome into a single refSNP. Newly submitted variations can either co-locate to form a new refSNP cluster, or may instead cluster with an already existing refSNP. When newly submitted variations cluster among themselves, they are assigned a new refSNP number, and when they cluster with an existing refSNP, they are added to that refSNP cluster.
Sometimes an existing refSNP will co-locate (identical coordinates) with another refSNP when dbSNP improves its clustering algorithm, when submissions are corrected, or when genome assemblies change between dbSNP builds. When existing refSNPs co-locate, the refSNP(s) with higher refSNP number(s) are retired (never to be reused), and all the submitted SNPs from the retired cluster(s) are re-assigned to the retained refSNP. The re-assignment of the submitted SNPs from a higher refSNP number to a refSNP cluster with a lower refSNP number is called a “merge,” and occurs during the “rs merge” step of the dbSNP mapping process. Merging is only used to reduce redundancy in the catalog of rs numbers so that each position has a unique identifier. All "rs merge" actions that occur are recorded and tracked.
Note: Originally, refSNPs clusters included variations of different class types because the submitted variations happen to map to the same location (true SNP, indels, mixed). dbSNP found that due to ballooning data submissions, however, refSNPs were becoming increasingly difficult to interpret because of the multiple variation class types present in each cluster. Since different variation classes represent different genetic events, dbSNP has since altered refSNP clusters to include only a single variation class type.
refSNP Splitting
Due to assembly changes or software updates, submissions previously calculated to be in the same cluster can be differentiated. In such cases, dbSNP separates or “splits” the cluster into two or more refSNP clusters depending on the particular circumstance. dbSNP may also split a cluster if newly submitted evidence indicates that two or more variation classes are clustered within a single refSNP number.
When an existing refSNP is split, those submitted SNP (ss) numbers that were most recently added to the cluster will be “split away” to form a new cluster. When this happens, the remaining ss numbers in the original cluster retain the old rsID number, while the ss numbers that are “split away” either cluster to another existing refSNP if they map to it, or are assigned a new refSNP number. The number of clusters that emerge from a split depends upon the number of distinct locations and types of variation classes that can now be identified.
RefSNP Number Stability
If a refSNP number has been merged into or split away from another refSNP number, it is very easy to use a retired refSNP number to find the current one (see hint below) . In other words, a refSNP number can be termed stable because merged or split refSNP numbers can always be traced to a previous refSNP number.
| Hint: |
|---|
There are three ways the you can locate the partner numbers of a merged refSNP, and one way to locate the partner of a split refSNP:
|
If, however, what is meant by “stable” is that the refSNP number of a particular variation always remains the same, then one should not consider a refSNP entirely stable, as a refSNP number may change if two refSNP numbers merge or split. Merging can occur if new evidence suggests that two refSNPs at a single sequence location are of the same variation type, and splitting will occur if mixed variation classes (e.g., SNV and indel) are clustered in a single refSNP. For more detail on merging and splitting, see the “Re-Mapping and RefSNP Merging” section and the “refSNP splitting” sections above.
A refSNP number may also change if:
- All of the submitted SNP (ss) numbers in a refSNP cluster are withdrawn by the submitter.
- dbSNP breaks up an existing cluster and re-instantiates a retired rs number based on a reported conflict from a dbSNP user.
Suspect Variations
Currently, a variant is flagged as a “suspect”, i.e., a potential false positive when the presence of a paralogous sequence in the genome (1,2) could cause mapping artifacts or if there is evidence suggesting sequencing errors or computation artifacts.
dbSNP will be updating its suspect false positive flagging system in the near future to rank suspect variants according to the amount of supporting evidence available for each refSNP. Those refSNP clusters that are suspect, but have data from multiple submitters indicating a heterozygous state exists, will rank more highly in dbSNP’s new system as a variation to be trusted than a suspect refSNP that has data from a single submission, has multiple submissions that show no evidence of heterozygosity, or with conflicting evidence of heterozygosity.
Molecular Class
dbSNP computes a molecular context for sequence variations by inspecting the flanking sequence for gene features during the contig annotation process, and does the same for RefSeq/GenBank mRNAs.
dbSNP has adopted Sequence Ontology (SO) terms to define its variation molecular classes so as to conform to the standard set by the biological community. The subset of SO terms dbSNP uses as functional classes can be found in Table 4.
Table 4.
Molecular codes for refSNPs in gene features
A variation may have multiple functional classes. Multiplicity will result, for example, when a variation falls within an exon of one transcript and an intron of another for the same gene.
Clinical Assertions
Novel variations and experimental evidence supporting clinical assertions (Table 5) are submitted to ClinVar. dbSNP and ClinVar will continue to support the Human Variation Batch Submission site as a Web-based tool that can be used to submit or update medically important variation submissions.
Table 5.
Clinical Significance organizes submissions by clinical assertion type
When novel variants with supporting clinical evidence are received from ClinVar, dbSNP remaps the asserted positions of the variants to the corresponding coordinates on the current assembly, as well as to cDNA, protein, and RefSeqGene sequences. Once the variants are mapped, dbSNP assigns ssIDs and rsIDs to each.
Data submitted through the Human Variation Batch Submission site that supports a clinical assertion are processed and extracted by ClinVar, which uses the data to assign clinical attributes to novel variants or to update the clinical attributes of existing variants (LSDB).
Once the submitted variants are mapped and their attributes assigned, these data are made available to other NCBI resources, including VarView, ClinVar, and Variation Reporter.
Population Diversity Data
Average Heterozygosity
The best single measure of a variation's diversity in different populations is its average heterozygosity. This measure serves as the general probability that both alleles are in a diploid individual or in a sample of two chromosomes. Estimates of average heterozygosity have an accompanying standard error based on the sample sizes of the underlying data, which reflects the overall uncertainty of the estimate. dbSNP’s computation of average heterozygosity and standard error for RefSNP clusters is available online. Please note that dbSNP computes heterozygosity based on the submitted allele frequency for each variation. If the frequency data for a variation is not submitted, dbSNP cannot compute the heterozygosity value, and therefore the refSNP report will show no heterozygosity estimate.
Additional population diversity data calculated for refSNP records includes population counts, individuals sampled for a variation, genotype frequencies, and Hardy Weinberg probabilities.
Minor Allele Frequency (MAF)
Minor Allele Frequency is the allele frequency for the 2nd most frequently seen allele. dbSNP aggregates the minor allele frequency for each refSNP cluster over multiple submissions to help users distinguish between common polymorphisms and rare variants.
Consider a variation with the following alleles and allele frequencies:
Reference Allele = G; frequency = 0.600
Alternate Allele = C; frequency = 0.399
Alternate Allele = T; frequency = 0.001
Based on the MAF guideline mentioned above, the minor allele is "C," so the minor allele frequency (MAF) is 0.399. Allele "T" with frequency 0.001 is considered a rare allele rather than a minor allele.
1000 Genomes Minor Allele Frequency (1000G MAF)
"1000G MAF" is the minor allele frequency (see above) based on genotype data from the 1000 Genomes Project [phase 1] global population of 1094 individuals.
Build Integration
dbSNP annotates the non-redundant set of variations (refSNP cluster set) on reference genome genomic sequences, chromosomes, mRNAs, and proteins as part of the NCBI RefSeq project. dbSNP computes summary properties for each refSNP cluster, which are then used to build fresh indexes for dbSNP in the Entrez databases, and to update the variation map in the NCBI Map Viewer. Finally, dbSNP updates links between dbSNP and BioProject, dbVar, dbGaP, Gene, HomoloGene, Nucleotide, OMIM, Protein, PubChem Substance, PubMed, PubMed Central, VarView, and Variation Reporter.
Public Release
Public release of a new build involves an update to the public database and the production of a new set of files on the dbSNP FTP site. dbSNP makes an announcement to the dbsnp-announce mailing list when the new build for an organism is publicly available.
The dbSNP Redesign: Changes to Clustering
As of this writing, dbSNP is planning a redesign that will introduce a number of fundamental changes to dbSNP’s dataflow. Users are encouraged to review the proposed changes below and to submit any comments and suggestions to vog.hin.mln.ibcn@nimda-pns. Among these changes is a new clustering algorithm.
While improvements continue to be made in genome assemblies, artificial sequence duplications will resolve and collapse, artificially collapsed regions of a genome will expand, and missing sequence regions will be added. In the case of a sequence collapse, the number of hits that a variation has on the assembly may be reduced, while in the case of a sequence expansion or addition, the number of hits that a variation has to the genome may increase.
In order to have the flexibility necessary to deal with these genome assembly changes, dbSNP 2.0 will have a new clustering algorithm that will change the concept of a refSNP as we know it. This new clustering algorithm will change the rules that govern clustering to invert the ss to rs relationship as it currently exists.
At present, multiple ss numbers can exist in the same rs cluster and each ss number in that cluster links to its one corresponding rs number. The new clustering rules, however, will change this relationship in that each rs will represent a unique location. Under these new rules, a single ss number could link to multiple rs numbers if the ss number maps to more than one location.
Access
The SNP database can be queried directly from the search bar at the top of the dbSNP homepage , by using the links to dbSNP resources and search options located on the homepage, or by accessing related NCBI resources that link to dbSNP data.
dbSNP Home Page
dbSNP is a part of the Entrez integrated information retrieval system and may be searched either by using an ID number query, or by using combinations of different search fields and qualifiers.
Single Record Query
Use the search bar at the top of the dbSNP homepage to find variations using dbSNP record identifiers. The record identifiers currently supported for single record queries are the reference SNP (refSNP) cluster ID number (rs#), the submitted SNP accession number (ss#), and the local (or submitter) ID number.
Complex Entrez Query
Use the SNP Advanced Search Builder page to construct a complex search using combinations of different search fields and qualifiers. The Advanced Search Builder allows you to construct a query by selecting multiple search terms from a large number of fields and qualifiers. See the Advanced Search Builder video tutorial for information about how to find existing values in fields and combine them to achieve a desired result.
dbSNP Batch Query
dbSNP Batch Query allows you to query using variation IDs (rs ID, ss ID, or local IDs), collected in a primary search to retrieve a large quantity of variations at the same time in a selected report format. Available report formats include ASN.1, BED, chromosome, FASTA, Flat File, genotype report, rs cluster report, ss detail report, and XML.
Variation Reporter
Variation Reporter matches submitted variation calls to variants housed in dbSNP or dbVar, thereby allowing access via a Web search or through an application programming interface (API) to all data and metadata that dbSNP has for the matching variants. If you submit novel variants and there are no matches between your data and the variants housed in dbSNP or dbVar, the Variation Reporter will provide the predicted consequence of each submitted variant.
BLAST
BLAST can be used to match submitted variations with asserted positions to matching dbSNP records (See the instructions at: ftp://ftp.ncbi.nih.gov/pub/factsheets/HowTo_Finding_SNP_by_BLAST.pdf. Query BLAST with the sequence or clone that contains the asserted position of the variant, and then select an appropriate reference database as the BLAST target. The BLAST algorithm will find any existing SNP records that map to the queried sequence, and thence to the variant of interest, if a dbSNP record happens to match it.
Note: If BLAST fails to find a matching dbSNP record for a variation of interest on a queried sequence:
- 1.
You cannot assume that the variant is novel without further study, because there are several reasons why an existing variant may not yet have a dbSNP record:
- a.
The sequence location of the existing variant may be missing from the reference assembly, or the transcript location of the variant has not yet been sequenced.
- b.
The existing variant may have been submitted with low sequence quality or ambiguous base calls, which would inhibit placement on the reference assembly.
- c.
The variant may exist in the literature, and has not yet been submitted by the author for inclusion in dbSNP. This is particularly true for those variants that were reported in historic literature.
- 2.
You can use Variation Reporter to get a predicted consequence of human variations to help you in your analysis if the variations have known sequence locations.
SNP Submission Information Queries
If refSNP(rs) or submitted SNP (ss) numbers are not available to use in a search for a dbSNP record, use the “Submission Information” module to construct a query that will select dbSNP variation records based on other available information associated with a submitted variation:
- Information associated with the submitter
- Information about the submitted batch that contains the variation of interest
- Information associated with the method used to assay for the variation (Table 2)
- Information associated with the submitted population
- Information associated with the publication reporting the variant
Search via ClinVar, Gene, or PubMed
There are multiple databases in NCBI that maintain links to dbSNP. Related records in dbSNP can be identified by following the Find related data on the Summary display, or following the links in the Related information section of a single record.
Entrez Programming Utilities (Eutils)
Use Entrez Programming Utilities (E-utilities or Eutils) to query dbSNP and retrieve information via Web services. You can test an Entrez query interactively and then execute that query using Eutils. There are a number of available Eutil programs that cover a wide range of query types. See the Entrez Programming Utilities help documentation for more information.
dbSNP FTP Site
NCBI supports the public distribution of dbSNP data by providing compressed data dumps in a number of different formats. Access to the NCBI FTP site is available via the World Wide Web (ftp://ftp.ncbi.nih.gov/snp/) or anonymous FTP (host ftp.ncbi.nih.gov cd snp). In addition to the data formats described on the FTP README file, which include ASN.1, FASTA, and XML, dbSNP FTP offers two additional formats:
VCF Format
The Variant Call Format, or VCF, was developed for the 1000 Genomes Project as a standardized format for storing large quantities of sequence variation data (SNPs, indels, larger structural variants, etc.) and any accompanying genotype data and annotation. A VCF file contains a header section and a data table section. Since the metadata lines in the header section can be altered to fit the requirements of the data to be submitted, you can use VCF to submit many different kinds of common variations (as well as their associated genotypes and annotation) that are contained within one reference sequence. VCF files are compressed (using bgzip) and are easily accessed. See Danecek, et. al. for a concise overview of VCF (3), and the official 1000 Genomes site for a detailed description of the VCF format. Submissions to dbSNP currently use VCF format version 4.1.
BED Format
The Browser Extensible Data (BED) format was developed by UCSC Genome Bioinformatics as a means of displaying data lines for genome browser annotation tracks. Each line of the BED format represents a single annotated feature that is described using required and optional fields. dbSNP BED files are derived from dbSNP RS Docsum ASN.1 (ftp://ftp.ncbi.nih.gov/snp/specs/docsum_3.4.xsd) and use the three required fields found in the standard BED format as well as three of the nine optional fields (name, score, strand). The dbSNP BED format has been QA tested and is compatible with standard BED tools and genome browser uploads such as the NCBI Remap Service (http://www.ncbi.nlm.nih.gov/genome/tools/remap), the UCSC Genome browser (https://genome.ucsc.edu/cgi-bin/hgGateway), and the EBI Genome Browser (http://www.ensembl.org).
ADA Section 508-Compliance Link
All links provided on the dbSNP homepage are also provided in text format at the bottom of the page to support browsing by text-based Web browsers. Suggestions for improving database access by disabled persons should be sent to the dbSNP development group at snp-admin@ncbi.nlm.nih.gov.
Local Copies of dbSNP
If you wish to create a SQL copy of dbSNP on a local server for direct access, use the directions in Appendix 3 of this chapter to create the tables and indices for dbSNP from the dbSNP schema, data, and SQL statements.
Note: We will be phasing out the relational database architecture of dbSNP during the dbSNP redesign, and are considering replacing it with Service Oriented Architecture (SOA) and a BLOB/CLOB store system in dbSNP 2.0. Storage technology and object schemas, however, are still under design. Since dbSNP 2.0 may not be an SQL based system, we will provide users with an API to access bulk dumps of data for those wanting to create a local copy of dbSNP. Check or subscribe to the dbSNP News and Announcements site for updates regarding the redesign and availability of the data as relational tables or as objects.
Related Tools and Studies
There are multiple tools related to processing or learning more about short sequence variations. These are described in depth in the variation overview section of the Handbook. In brief, they support the following use cases:
Converting a Location on One Assembly or Sequence to Another
NCBI's Genome Remapping Service (Remap) allows you to convert locations from one sequence to another based on alignments. Use Remap if you have identified the location of variation on an assembly, or on a RefSeqGene/LRG, and want to determine the location on a different assembly (or on the genome in the case of the RefSeqGene).
History of Interpretation of the Medical Importance of an Allele
ClinVar archives the relationship reported between variations and phenotype by accessioning and versioning submissions.
Association Studies
dbGaP archives and distributes data from studies that examine the relationship between phenotype and genotype. Such studies include Genomewide Association Studies (GWAS), medical sequencing, and molecular diagnostic assays. Links are available from dbGaP controlled access records to related variation data in dbSNP, but there are no reciprocal links from dbSNP records to dbGaP unless the aggregate data are public. The refSNP report “Association” section will link to association results from the NHGRI GWAS Catalog and/or PheGenI when association data are available.
Histocompatibity
dbMHC provides a platform where users can access, submit, and edit data related to the human Major Histocompatibility Complex, also called the HLA (Human Leukocyte Antigen).
Both dbMHC and dbSNP store the underlying variation data that define specific HLA alleles. dbMHC provides access to related dbSNP records at the haplotype and variation level, whereas dbSNP provides access to related dbMHC records at the haplotype level.
Haplotypes
The International HapMap Project site allows access to its catalog of statistically related variations, also known as haplotypes, for a number of different human populations, and is a useful resource for those researchers looking for variations associated with a particular gene. HapMap haplotypes can be searched by a landmark such as a refSNP number or gene symbol, as well as by sequence region or chromosome region. The resulting HapMap report includes an ideogram with various tracks that can be altered to provide required data, and appropriate tracks in the report will provide direct links to refSNP cluster records.
Variation as Related to Citations, Genes, Phenotypes, and other NCBI Databases
Multiple databases in NCBI can be used to identify variation that meets certain criteria. They may either reference rs numbers explicitly, or provide links from their records to records in dbSNP.
Variation Batch Submission (VarBatch)
VarBatch is an online submission resource for both clinical and non-clinical human variations, and allows the update and annotation of previously submitted variations. When an asserted clinical variation is processed through VarBatch, it is assigned both a dbSNP submitted SNP (ss) accession as well as a ClinVar accession (format: SCV000000000.0), since the ClinVar accession represents the asserted variation/phenotype relationship.
Note: Since VarBatch does not accept frequency, genotype, or population data, submit these data to dbSNP as updates to your VarBatch submission using the dbSNP VCF or Flat File format via email or through a pre-arranged FTP upload once ss numbers are assigned to your submitted variations.
Variation Reporter
Variation Reporter matches submitted variation call data to variants housed in dbSNP or dbVAR, allowing access to all data and metadata that dbSNP has for any known matching variants. If you submit novel variants to the Variation Reporter, and there are no matches between your data variants housed in dbSNP or dbVAR, the Variation Reporter will provide the predicted consequence of each submitted variant.
VarView
VarView reports display detailed variation information associated with a particular gene and are created only for those genes that have asserted clinical variations. VarView can be accessed in two ways:
- 1.
Through Gene by using the query “gene_snp_clin[filter]” to identify gene records that have a VarView report.
- 2.
Through dbSNP either by using the “VarView” link
 displayed in refSNP reports for variations that have asserted clinical significance, or by querying dbSNP using “snp_gene_clin[filter]” to identify variants that have a VarView report.
displayed in refSNP reports for variations that have asserted clinical significance, or by querying dbSNP using “snp_gene_clin[filter]” to identify variants that have a VarView report.
Once a Gene or dbSNP record has been selected, and the VarView link on the record has been activated, a VarView report will appear that includes:
- A brief description the gene
- A list of all observed rs variants of the gene
- Links to both internal and external resources including locus specific databases (LSDB), OMIM, Gene, and PubMed.
When one of the listed rs variations in the VarView report is selected, the “submission details” section of the report provides a list of ss numbers associated with the selected rs number as well as links to submitter sites and each ss report.
Note: VarView will be replaced by a new Variation Gene Viewer in April 2014. This new resource will allow users to access all of NCBI’s variation data (i.e., dbSNP, dbVar, ClinVar) in a gene-centric fashion.
1000 Genomes Browser
The 1000 Genomes Browser provides access to 1000 Genomes data including variations, genotypes, and sequence read alignments within the context of GRCh37, the reference assembly used by the 1000 Genomes Project for analysis. The browser allows you to configure the display to include multiple data tracks of interest and provides links to related data housed in various NCBI resources. The 1000 Genomes Browser allows users to quickly view alignments supporting a particular variant call and can be used to download and read variant data for small genomic regions of interest.
Access the 1000 Genomes Browser from dbSNP using the 1000 Genomes Browser link  in the refSNP report “Integrated Maps” section.
in the refSNP report “Integrated Maps” section.
Access dbSNP from the 1000 Genomes Browser using the “hover” feature in either the “Clinical Channel” or “Cited Variant” tracks. Click on the variation rsID that appears.
References
- 1.
- Musumeci L, Arthur JW, Cheung FS, Hoque A, Lippman S, Reichardt JK. Single nucleotide differences (SNDs) in the dbSNP database may lead to errors in genotyping and haplotyping studies. Hum Mutat. 2010 Jan;31(1):67–73. [PMC free article: PMC2797835] [PubMed: 19877174]
- 2.
- Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J. 1000 Genomes Project, Eichler EE. Diversity of human copy number variation and multicopy genes. Science. 2010 Oct 29;330(6004):641–6. [PMC free article: PMC3020103] [PubMed: 21030649]
- 3.
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R. 1000 Genomes Project Analysis Group. The variant call format and VCFtools. Bioinformatics. 2011 Aug 1;27(15):2156–8. [PMC free article: PMC3137218] [PubMed: 21653522]
Appendices
Appendix 1. dbSNP Report Formats
ASN.1
The docsum_3.4.asn file is the ASN structure definition file for ASN.1 and is located in the /specs subdirectory of the dbSNP FTP site. The 00readme file, located in the main dbSNP FTP directory, provides information about ASN.1 data structure and data exchange. ASN.1 text or binary output can be converted into one or more of the following formats: Flat File, FASTA, DocSum, Chromosome Report, RS/SS, and XML.
Note: ASN.1 data must be retrieved programmatically by using eUtils or by using the dbSNP Batch Query Service.
BED
The Browser Extensible Data (BED) format was developed by UCSC Genome Bioinformatics as a means of displaying data lines for genome browser annotation tracks.
Each line of the BED format represents a single annotated feature that is described using required and optional fields. dbSNP BED files are derived from dbSNP RS DocSum ASN.1 (ftp://ftp.ncbi.nih.gov/snp/specs/docsum_3.4.xsd) and use the three required fields found in the standard BED format as well as three of the nine optional fields (name, score, strand).
The dbSNP BED format has been QA tested and is compatible with standard BED tools and genome browser uploads such as the NCBI Remap Service (http://www.ncbi.nlm.nih.gov/genome/tools/remap), the UCSC Genome browser (https://genome.ucsc.edu/cgi-bin/hgGateway), and the EBI Genome Browser (http://www.ensembl.org).
Chromosome Report
The Chromosome Reports format provides an ordered list of RefSNPs in approximate chromosome coordinates and contains a great deal of information about each variation. Since the coordinate system used in this format is the same as that used for the NCBI Genome Map Viewer, Chromosome Reports contains information helpful in the identification of variations that can be used as markers.
A full description of the information provided in the Chromosome Reports format is available in the 00readme file, located in the SNP directory of the SNP FTP site.
Note: A Chromosome Report’s directory may contain any of the following files:
- chr_AltOnly.txt.gz: List of variations that map to a non-reference (alternate) assembly (e.g., a human refSNP maps to HuRef or TCAGChr7, but not to GRC)
- chr_MT.txt.gz: List of variations that map to the mitochondria chr_Multi.txt.gz: List of variations that map to multiple chromosomes
- chr_NotOn.txt.gz: List of variations that did not map to any chromosomes
- chr_PAR.txt.gz: List of variations on the pseudoautosomal regions of human or great ape X and Y chromosomes.
- chr_UN.txt.gz: List of mapped variations that are on unplaced chromosomes
FASTA: ss and rs
The FASTA report format provides the flanking sequence for each report of variation in dbSNP, as well as for all the submitted sequences that have a report of “no variation.” The FASTA data format is typically used for sequence comparisons using BLAST.
Online BLAST is useful for conducting a few sequence comparisons in the FASTA format, whereas multiple FASTA sequence comparisons require the installation of a local stand-alone version of BLAST, and the construction of a local database of FASTA formatted data.
A full description of the information provided in the FASTA report format is available in the 00readme file, located in the SNP directory of the SNP FTP site.
Gene Report
The dbSNP Gene report is a text report that provides a list of all refSNPs currently known to be located in a particular gene, as well as a summary of general and clinical information for each listed variation. The file naming convention for gene_report is “XXXXX_gene_ report.txt.gz,” where “XXXX” represents the gene symbol (e.g., LPL, the gene symbol for lipoprotein lipase).
A full description of the information provided in the gene_ report format is available in the 00Gene_report_format_README, located in the human gene_report directory of the of the SNP FTP site.
Genotype Report
Since the massive amount of genotype data we receive from large sequencing projects (e.g.,1000 Genomes) makes it difficult for NCBI to maintain and query the dbSNP SQL tables, we will no longer provide genotype data or reports.
NCBI is currently developing a new service (Genotype Server) that will more efficiently store and serve genotype and frequency data using API, the internet, and FTP. It should be available sometime in 2014.
The genotype XML, on the dbSNP FTP server, is still available and provides submitter and genotype information for many submitted SNPs. It is organized in chromosome specific files under each organism directory in the “genotype subdirectories” (e.g., human genotype XML files are located in ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606/genotype/). Users should be aware, however, that the genotype XML is also in the process of being phased out.
Note: Until NCBI’s new Genotype Server is released, genotype data can be queried and downloaded at these two alternative sites:
1000 Genomes: http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/
HapMap: http://hapmap.ncbi.nlm.nih.gov/
rs docsum flatfile
The rs docsum flatfile report is generated from the ASN.1 datafiles and is provided in the files whose naming convention is "/ASN1_flat/ds_flat_chXX.flat". Files are generated per chromosome (chXX in file name), as with all of the large report dumps.
Because flatfile reports are compact, they will not provide you with as much information as the ASN.1 binary report, but are useful for manual scanning of human SNP data because they provide detailed information at a glance.
A full description of the information provided in the rs docsum flatfile format is available in the 00readme file, located in the SNP directory of the SNP FTP site.
VCF
The Variant Call Format, or VCF, was developed for the 1000 Genomes Project as a standardized format for storing large quantities of sequence variation data (SNPs, indels, larger structural variants, etc.) and any accompanying genotype data and annotation.
A VCF file contains a header section and a data table section. Since the metadata lines in the header section can be altered to fit the requirements of the data to be submitted, you can use VCF to submit many different kinds of common variations (as well as their associated genotypes and annotation) that are contained within one reference sequence. VCF files are compressed (using bgzip), and are easily accessed.
See Danecek, et. al. for a concise overview of VCF (3) and the official 1000 Genomes site for a detailed description of the VCF format. Submissions to dbSNP currently use VCF format version 4.1.
XML
The XML format provides query-specific information about refSNP clusters, as well as cluster members in the NCBI SNP Exchange (NSE) format. The XML schema is located in the docsum_3.4.xsd file, which is housed in the /specs subdirectory of the dbSNP FTP site. A human-readable text form of the NSE definitions can be found in docsum_3.4.asn, also located in the /specs subdirectory of the dbSNP FTP site.
Note: XML data must be retrieved programmatically by using eUtils or by using the dbSNP Batch Query Service.
Appendix 2. Rules and Methodology for Mapping
The appearance of FASTA-formatted genome sequence for a new build of an assembly or the significant accrual of newly submitted SNP data for an organism will initiate a cycle of MegaBLAST and BLAST alignment of the variations to the NCBI genome assembly of the organism.
Variation Placement by Remapping
dbSNP uses sequence alignments to map asserted locations and underlying features on to reference sequences. During the build process, dbSNP performs three types of remapping: Mapping up, Mapping Down, and Assembly to Assembly remapping.
Mapping Up
“Mapping Up” refers to the process of mapping a submitted variation whose location is based on a reference sequence, cDNA, or protein to the current genome build and to RefSeqGene using sequence alignments.
Mapping up from cDNA to Genomic Sequence
If the provided location is in an exon, dbSNP maps the input coordinates directly to the genome through available alignments. If the provided location is in an intron, dbSNP maps the exon boundary coordinate that is closest to the intron position, again using available alignments.
Mapping up from Protein to cDNA
dbSNP aligns the protein accession and location as well as the asserted location of the variation on the protein to cDNA. This alignment generates up to three possible sequence locations of the variation at the nucleotide level, where it is possible to discern the stated variation at the protein level.
Mapping Down
“Mapping Down” refers to the process of using genomic alignments to map information found on a genomic sequence to transcript sequences and proteins.
Assembly to Assembly Remapping
Assembly-Assembly remapping allows the projection of features from one assembly coordinate system to another using genomic alignments. dbSNP performs a base by base analysis of each feature on the source sequence in order to project the feature through the alignment to the new sequence.
Variation Placement by BLAST
When an asserted location for a submitted variant is not available, dbSNP will attempt to place the variation on the genome by BLASTing submitted variation flanking sequences against a genomic assembly. This mapping process is a multi-step, computer-based procedure that begins when refSNP and submitted SNP FASTA sets are aligned to the most recent genome assembly using BLAST or MegaBLAST. The quality of each alignment is determined using an Alignment Profiling Function.
The BLAST/MegaBLAST output of the ASN.1 binary files of local alignments is analyzed by an algorithm to create a group of local alignments that lay close to one another on a sequence. If the global alignment is greater than or equal to a pre-determined percentage of the flanking sequence, it is accepted as a true alignment between the refSNP or submitted SNP and the genome assembly.
This group of close local alignments is then processed to define alleles and LOC types for each hit and to establish the hit location. The output is filtered to remove paralogous hits and to select those variations that have the greatest degree of alignment to a particular contig. The output is then placed into a file and processed to create an MD5 positional signature for each variation. These signatures are then placed in the SNP MAP INFO file and loaded into dbSNP.
Once all the results from previous steps are loaded into dbSNP, dbSNP looks for clustering candidates. If an MD5 signature for a particular SNP is different from the MD5 signature of another SNP, then each SNP will have a unique hit pattern and need not be clustered. If an MD5 signature of a particular SNP is the same as that of another SNP, the two SNPs may have the same hit pattern, and if after further analysis, the hit patterns are shown to be the same, the two SNPs will be clustered.
Appendix 3. How to Create a Local Copy of dbSNP
How to Create a Local Copy of dbSNP
Curently, dbSNP is a relational database that contains hundreds of tables. Since the inception of build 125, the design dbSNP has been altered to a ”hub and spoke” model, where the dbSNP_Main_Table acts as the hub of a wheel, storing all of the central tables of the database, while each spoke of the wheel is an organism-specific database that contains the latest data for a specific organism. dbSNP exports the full contents of the database for the public to download from the dbSNP FTP site. During the dbSNP redesign, however, we will be phasing out the relational database architecture of dbSNP, and are considering replacing it with Service Oriented Architecture (SOA) and a BLOB/CLOB store system in dbSNP 2.0.
Due to security concerns and vendor endorsement issues, dbSNP cannot provide users with direct dumps of dbSNP. The task of creating a local copy of dbSNP can be complicated and should be left to an experienced programmer. The following sections will guide you in the process of creating a local copy of dbSNP, but these instructions assume knowledge of relational databases, and were not written with the novice in mind.
If you have problems establishing a local copy of dbSNP, please contact dbSNP at vog.hin.mln.ibcn@nimda-pns.
Schema: The dbSNP Physical Model
A schema is a necessary part of constructing your own copy of dbSNP because it is a visual representation of dbSNP that shows the logical relationship between the data. It is available as a printable PDF file from the dbSNP FTP site.
Data in dbSNP are organized into “subject areas” depending on the nature of the data. The data dictionary includes a description of the tables in dbSNP as well as tables of columns and their properties. Foreign keys are not enforced in the physical model because they make it harder to load table data asynchronously. In the future, dbSNP will add descriptions of individual columns. The data dictionary is also available online from the dbSNP website.
Resources Required for Creating a Local Copy of dbSNP
Software:
- Relational database software. If you are planning to create a local copy of dbSNP, you must first have a relational database server, such as Sybase, Microsoft SQL server, or Oracle. dbSNP at NCBI runs on an MSSQL server version 2000, but there are users who have successfully created their local copy of dbSNP on Oracle.
- Data loading tool. Loading data from the dbSNP FTP site into a database requires a bulk data-loading tool, which usually comes with a database installation. An example of such a tool is the bcp (bulk-copy) utility that comes with Sybase, or the “bulkinsert” command in the MSSQL server.
- winzip/gzip to decompress FTP files. Complete instructions on how to uncompress *.gz and *.Z files can be found on the dbSNP FTP site.
Hardware:
- Computer platforms/OS
Databases can be maintained on any PC, Mac, or UNIX with an internet connection.
- Disk space
To ascertain the disk space needed for a complete copy of dbSNP for a particular organism, determine the total download file size for the organism as a starting point. You need a minimum of three times of the data file size to have space for creating indices and storing your own working tables. The allocated size of dbSNP human B137 on dbSNP’s internal server is 3TB, while mouse B137 size is about 700GB.
- Memory
The minimum amount of memory required is approximately 4GB.
- Internet connection
dbSNP recommends a high-speed connection to download such large database files.
dbSNP Data Location
The FTP database directory in the dbSNP FTP site contains the schema, data, and SQL statements to create the tables and indices for dbSNP:
- The shared_schema subdirectory contains the schema DDL (SQL Data Definition Language) for the dbSNP_main_table.
- The shared_data subdirectory contains data housed in the dbSNP_main_table that is shared by all organisms.
- The organism_schema subdirectory contains links to the schema DDL for each organism specific database.
- The organism_data subdirectory contains links to the data housed in each organism specific database. The data organized in tables, where there is one file per table. The file name convention is: <tablename>.bcp.gz. The file name convention for the mapping table also includes the dbSNP build ID number and the NCBI genome build ID number. For example, B125_SNPContigLoc_35_1 means that during dbSNP build 125, this SNPContigLoc table has SNPs mapped to NCBI contig build 35 version 1. The data files have one line per table row. Fields of data within each file are tab delimited.
dbSNP uses standard SQL DDL(Data Definition Language) to create tables, views for those tables, and indexes. There are many utilities available to generate table/index creation statements from a database.
| Hint |
| If your firewall blocks passive FTP, you might get an error message that reads: “Passive mode refused. Turning off passive mode. No control connection for command: No such file or directory.” If this happens, try using a "smart" FTP client like NCFTP (available on most UNIX machines). Smart FTP clients are better at auto-negotiating active/passive FTP connections than are older FTP clients (e.g., Sun Solaris FTP). |
Stepwise Procedure for Creating a Local Copy of dbSNP
- 1.
Prepare the local area
(check available space, etc.)
- 2.
Download the schema files
- a.
Download the following files from the dbSNP shared
_schema subdirectory: dbSNP_main_table, dbSNP_main_index_constraint, and all the files in the shared _data subdirectory. Together, the files from both of these subdirectories will allow you to create tables and indices for the dbSNP_main_table. - b.
Go to the organism
_schema subdirectory and select the organism for which you wish to create a database. For the purpose of this example, human_9606 has been selected. Once human_9606 is selected, you will be directed to the human organism_schema subdirectory. Download all of the files contained in this subdirectory. - c.
Go to the organism
_data subdirectory, and select the organism for which you wish to create a database. For the purpose of this example, human_9606 has been selected. Once you select human_9606, you will be directed to the human organism_data subdirectory. Download all of the files contained in this subdirectory. A user must always down load the files located in the most recent versions of the shared_schema and shared_data subdirectories in addition to any organism specific content.
Save all the files in your local directory and decompress them.
Hint: On a UNIX operating system, use gunzip to decompress the files: dbSNP_main_table and dbSNP_main_index_constraint.
The files on the SNP FTP site are UNIX files. UNIX, MS-DOS, and Macintosh text files use different characters to indicate a new line. Load the appropriate new line conversion program for your system before using bcp.
- 3.
Create the dbSNP_main_table
- a.
From the shared
_schema subdirectory, use the dbSNP_main_table file to create tables, and use the dbSNP_main_index_constraint files to create indices for the dbSNP main database. - b.
Load all of the bcp files located in the shared
_data subdirectory into the dbSNP_main_table you just created using the data-loading tool of your database server (e.g., bcp for Sybase). See the sample FTP protocol and sample Unix C Shell script (below) for directions. - c.
Create indices by opening the dbSNP_main_index_constraint.sql file. If you are using a database server that provides the isql utility, then use the following command:
isql -S <servername> -U usename -P password -i dbSNP_main_index_constraint.sql
Hint: The “.bcp” files in the shared_data and organism_data subdirectories may be loaded into most spreadsheet programs by setting the field delimiter character to “tab”.
- 4.
Create the organism specific database
Once the dbSNP_main_table has been created, create the organism specific database using the files in your specific organism’s organism_schema and organism_data subdirectories. Human_9606 will be used for the purpose of this example:- a.
Create the human_9606 database using the following files found in the human_9606 organism
_schema: human_9606_table.sql.gz, human_9606_view.sql.gz, human_9606_index_constraint.sql.gz,
and human_9606_foreign_key.sql.gz- b.
Load all of the bcp files located in the shared
_data subdirectory into the human_9606 database you just created using the data-loading tool of your database server (e.g., bcp for Sybase). See the sample FTP protocol and sample Unix C shell script (below) for directions. Hint: Use “ftp -i” to turn off interactive prompting during multiple file transfers to avoid having to hit “yes” to confirm transfer hundreds of times. Hint: To avoid an overflow of your transaction log while using the bcp command option
(available in Sybase and SQL servers), select the "batch mode" by using the
command option: -b number of rows. For example, the command option -b 10000 will cause a commit to the table every 10,000 rows.
- 5.
Sample FTP Loading protocol
- a.
Type ftp -i ftp.ncbi.nih.gov (Use "anonymous" as user name and your email as your password).
- b.
Type: cd snp/database
- c.
To get dbSNP_main for shared tables and shared data: Type ls to see if you are in the directory with the right files. Then type “cd shared_schema” to get schema file for dbSNP_main, and finally, type “cd shared_data” to get the data for dbSNP_main.
- d.
Type binary (to set binary transfer mode).
- e.
Type mget *.gz (to initiate transfer). Depending on the speed of the connection, this may take hours since the total transfer size is gigabytes in size and growing.
- f.
To decompress the *.gz files, type gunzip *.gz. (Currently, the total size of the uncompressed bcp files is over 10 GB).
- 6.
Use scripts to automate data loading.
- a.
Located in the loadscript subdirectory of the dbSNP FTP site, there is a file called cmd.create_local_dbSNP.txt that provides a sample UNIX C shell script for creating a local copy of dbSNP_main and a local copy of a specific organism database using files in the shared_schema, and the organism_schema subdirectories.
- b.
Also in the the loadscript subdirectory of the dbSNP FTP site, there is a file called cmd.bulkinsert.txt that provides a sample UNIX C shell script for loading tables with files located in shared_data and organism_data subdirectories.
- 7.
Data integrity (creating a partial local copy of dbSNP)
dbSNP is a relational database. Each table has either a unique index or a primary key. Foreign keys are not reinforced. There are advantages and a disadvantage to this approach. The advantages are that this approach makes it easy to drop and recreate the table using the dbSNP_main_table, which then makes it possible to create a partial local copy of dbSNP. For example, if you are interested only in the original submitted SNP and their population frequencies, and not in their map locations on NCBI genome contigs or GenBank Accession numbers (both are huge tables), then these tables can be skipped (i.e., SNPContigLoc and MapLink). Please remember that mapping tables such as SNPContigLoc will have a build ID prefix and suffix included in its file name. (e.g., SNPContigLoc will be b125_SNPContigLoc_35_1for SNP build 125, and NCBI contig build 35 version 1). Of course, to select tables for a particular query, the contents of each table and the dbSNP entity relationship (ER) diagram need to be understood. The disadvantage of unreinforced references is that either the stored procedures or the external code needs to be written to ensure the referential integrity.
- The Database of Short Genetic Variation (dbSNP) - The NCBI HandbookThe Database of Short Genetic Variation (dbSNP) - The NCBI Handbook
Your browsing activity is empty.
Activity recording is turned off.
See more...