

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
SRA Handbook [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.
This publication is provided for historical reference only and the information may be out of date.

Overview
The purpose of this document is to explain to users how to download datasets of interest and associated metadata.
Important Notes on Download Facilities
- One basic format (.sra) is provided by the SRA for all publicly available data. The SRA Toolkit is provided to allow conversion to several popular formats.
- At a minimum, users are advised to use Aspera Connect (or the equivalent command line tool, ascp) for bulk downloads, rather than HTTP or FTP. Aspera provides faster bandwidth, a higher level of flow control, user level encryption, and the ability to download trees of components.
- We most strongly recommend the use of the SRA Toolkit to download data files directly. The individual utilities are able to resolve SRA accessions and initiate downloads automatically. The ‘prefetch’ utility is specifically provided for researchers that wish to download SRA data using a command line utility.
Related Documents
Notices
Reference herein to any specific commercial products, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government, and shall not be used for advertising or product endorsement purposes.
Software Version
This guide is current to SRA Toolkit version 2.5.x. Instructions for previous versions of the SRA Toolkit may be different from those provided in this guide. We recommend that users stay current with SRA Toolkit updates to benefit from feature additions and bug fixes.
Reference Compression
Compression by reference is a sequence alignment compression process for storing data. Compression by reference stores the difference in base pairs between sequence data and the segment(s) to which it is aligned. Throughout this document you will note that the behavior and properties of reference compressed SRA data and conventional data differ significantly. Notably,
- The SRA Toolkit can output reference-compressed data as aligned sam and can perform pileup analysis.
- The SRA Toolkit requires internet connectivity in order to download reference sequences in order to process aligned SRA data.
- Only aligned data can be viewed in the NCBI Sequence Viewer.
- Aligned data cannot be filtered in the SRA Run Browser.
- Aligned data cannot currently be searched in SRA BLAST (this is actively being developed).
Download with Prefetch
The SRA Toolkit can be used to directly download SRA data files and reference sequences (see the “Reference Compression” section above). We strongly encourage users to use these methods to access SRA data as they are simple to use and they avoid many of the manual steps required by other methods (searching FTP directories, browsing and clicking, etc.).
The SRA Toolkit will have to be properly configured in order to access NCBI servers and download data. Recent versions of the Toolkit are packaged with a ‘default’ configuration that should work for most users. Please review the pros and cons for using the default configuration here. If the default configuration does not work for your installation, or you wish to customize aspects of file handling by the Toolkit (e.g., where downloaded files are stored locally), you will need to configure the Toolkit and then test it to confirm that it is operating as expected. Please email vog.hin.mln.ibcn@ars if you have any problems configuring or using the Toolkit.
Prefetch
The ‘prefetch’ utility in the SRA Toolkit can be used to download SRA data and any required reference sequences in a single operation. Prefetch can use either HTTP (default) or ascp (if installed) to contact the SRA, resolve the accessions that you have specified, and then download the data. Prefetch can be used on single data files or to batch download several at a time. Below is an example prefetch command with the expected output. More information can be obtained on the prefetch documentation page and by executing ‘prefetch --help’.
$ prefetch SRR390728
Maximum file size download limit is 20,971,520KB
2016-01-14T16:57:02 prefetch.2.5.7: 1) Downloading 'SRR390728'...
2016-01-14T16:57:02 prefetch.2.5.7: Downloading via fasp...
2016-01-14T16:57:08 prefetch.2.5.7: fasp download succeed
2016-01-14T16:57:08 prefetch.2.5.7: 1) 'SRR390728' was downloaded successfully
2016-01-14T16:57:09 prefetch.2.5.7: 'SRR390728' has 25 unresolved dependencies
2016-01-14T16:57:09 prefetch.2.5.7: 2) Downloading 'ncbi-acc:GPC_000000394.1?vdb-ctx=refseq'...
2016-01-14T16:57:09 prefetch.2.5.7: Downloading via fasp...
2016-01-14T16:57:13 prefetch.2.5.7: fasp download succeed
2016-01-14T16:57:13 prefetch.2.5.7: 2) 'ncbi-acc:GPC_000000394.1?vdb-ctx=refseq' was downloaded successfully
2016-01-14T16:57:13 prefetch.2.5.7: 3) Downloading 'ncbi-acc:GPC_000000395.1?vdb-ctx=refseq'...
2016-01-14T16:57:13 prefetch.2.5.7: Downloading via fasp...
2016-01-14T16:57:15 prefetch.2.5.7: fasp download succeed
Note that the example file is reference-compressed and that prefetch automatically obtains the reference sequences required to extract data from the .sra file. If your Toolkit installation is not properly configured, or you elect to block the ability of the Toolkit to contact NCBI, you will then need to determine (1) if your downloaded dataset is reference-compressed, (2) if so, which references are required to access the data (see vdb-dump for an example of how to determine this), and (3) acquire the reference sequences manually here.
Other Toolkit utilities
All SRA Toolkit functions - most notably the ‘dump’ utilities that convert SRA data into other formats - are able to download data “on-the-fly” at runtime. This works like prefetch, as the tools will also automatically acquire all needed reference sequences. To invoke a Toolkit utility to download data as they are converted to your preferred format, simply execute the utility on an SRA accession rather than a local file. In other words, the command
$ fastq-dump --split-files SRR390728
Is implicitly requesting that fastq-dump download SRR390728 and its references from the SRA and then output the data in fastq format. Conversely,
$ fastq-dump --split-files ~/Downloads/SRA/SRR390728.sra
Is instructing fastq-dump to operate on a local file that was previously downloaded from the SRA. In this case fastq-dump would still attempt to contact NCBI to obtain the references needed to convert the data to fastq (unless you have specifically configured the Toolkit to not contact NCBI).
The Run Browser
The SRA Run Browser can display sequencing and instrumentation data on a given run. Typically the Run Browser is reached as a click through from an Entrez SRA Experiment report. Users may also navigate by entering a run accession (SRR, DRR, or ERR) directly in the Run Browser.
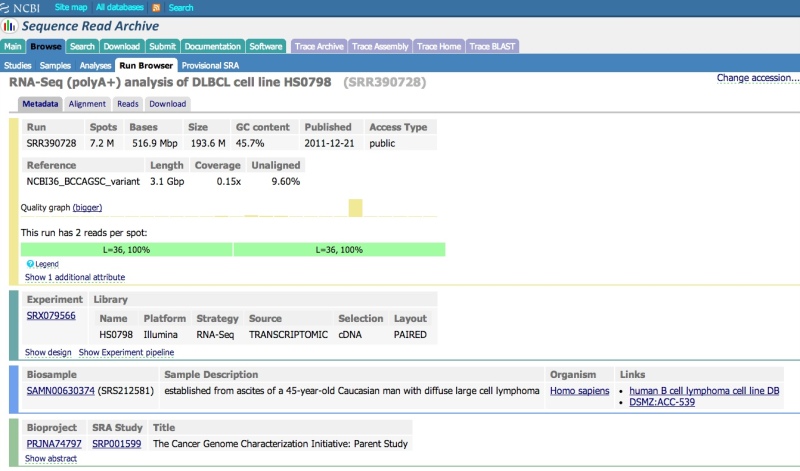
Viewing data in the Run Browser
Reference compressed (aligned) SRA data have an “Alignment” tab. Clicking on this tab will allow you to configure the NCBI Sequence Viewer to display the data aligned to a reference sequence.

To view the raw reads in a single Run, click on the “Reads” tab. Individual reads can be viewed and searched (see next section – note that only unaligned data can currently be searched). Various options can be applied using the “View” menu (e.g., display decimal quality scores, technical reads, etc.).
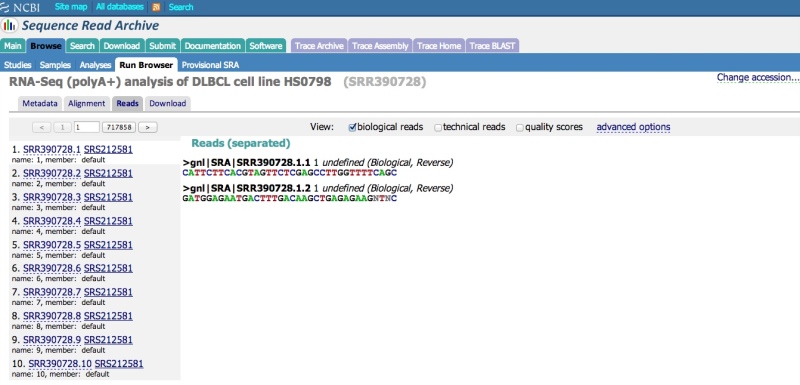
The Run Browser supports IUPAC single letter nucelotide codes (data submitted in color space are presented in base space; the SRA Toolkit can be used to download and output the data in color space, if required). Quality scores are presented in the Phred scale.
Filtering and Selection
The Reads tab in the Run Browser can be used to filter and search reads according to certain regular expression pattern matching:
- Sequence substring: one of the biological reads for a spot should contain the substring. Examples: ATTGGA, ^ATTGGA, ATTGGA$, ATGDNNAT, and ATGGA&GCGC. The strings are case insensitive, and belong to either 2NA or 4NA alphabets. String length limited to 29 characters in 4NA alphabet (includes IUPAC substitution codes) or 61 characters in 2NA alphabet (ACGT only). Search is case insensitive and strings may be combined with boolean operators & | ! (AND, OR, NOT). See "SRA nucleotide search expressions" for more details.
- Name of a spot you are looking for. Example: EXWA4RL02G9Z6H
- Name of sample pool member, or "all" for all members. Example: M22_V2 will return all spots assigned to the sample pool member M22_V2 for run SRR031989.
- Spot Id. Example: 23
Please note that the filter searches across read boundaries within each spot. Thus, pattern matches within technical reads and across paired-end data boundaries will also be returned.
The filter provided in the Run Browser has limited functionality, but is quite fast if you are looking to quickly search a single Run for a defined sequence of interest. Please see the section below on SRA BLAST if you require more advanced searching or searches across multiple sequencing libraries.
Downloading Data from the Run Browser
Clicking on the “Download” tab in the Run Browser will present a selection of links that will allow you to download (1) an individual dataset (Run), (2) all datasets in a given sequencing library (Experiment), or (3) all datasets linked to a given project (Study). You are also provided with three download choices: Aspera (using the Aspera Connect plugin), HTTP (using your browser), FTP (using command line FTP or a client).

SRA BLAST
SRA BLAST can be used to for advanced searching of single or multiple sequencing libraries from the same or different projects. There are two ways to access SRA BLAST in order to build a “search space” from which you are attempting to pull matches to your sequence(s) of interest. Successful BLAST searches will lead you to a results / summary page that can be used to download reads of interest or be directed to the SRA Run Browser to further investigate or download the entire dataset.
Note that SRA BLAST currently has a limit of 211 reads (approximately 2 billion) per search – attempts to add more than 211 reads will result in an error and rejection of the search. Users that require more substantial search capability are advised to contact the SRA (vog.hin.mln.ibcn@ars) to determine if other SRA BLAST tools might be of use.
Sending Entrez results to SRA BLAST
After performing an Entrez query to restrict results to datasets of interest, you may use the “Send to” feature to select datasets of interest and send them to SRA BLAST.
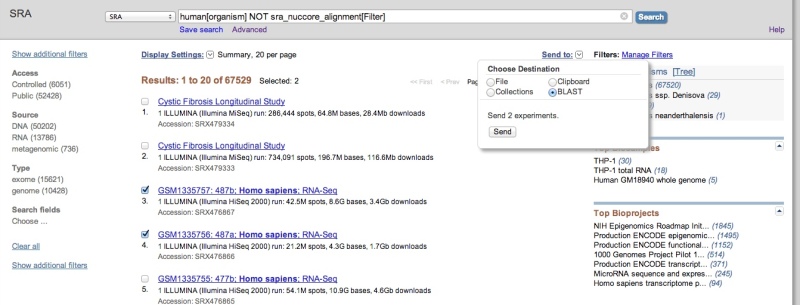
SRA-BLAST does not currently support reference compressed SRA datasets, so it is generally advised that you add the condition ‘NOT sra_nuccore_alignment[Filter]’ (as in the above example) to your queries to remove these datasets from the search results. Attempting to send incompatible datasets to BLAST will result in an error like the following:

If you believe that the data you are attempting to search against should be BLAST-able, but are not, please email vog.hin.mln.ibcn@ars for assistance and advice. After successfully sending accessions to SRA BLAST, you are then able to input your sequence(s) of interest and perform the search.
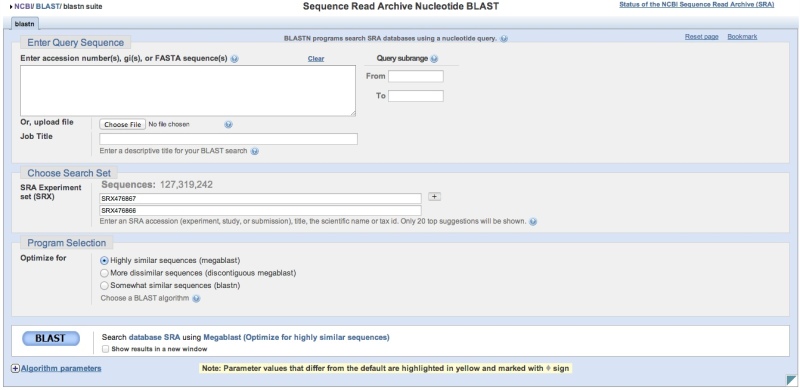
Building a search list in SRA BLAST directly
SRA BLAST can be accessed directly. You will then need to provide SRA Experiment (SRX, DRX, or ERX) accessions or use the autocomplete feature to help refine your search. You may enter 1 Experiment accession per line in the search list. The ‘+’ button can then be used to add additional sequencing libraries to the search space. Note that a running tally of the number of sequences is presented above the list of accessions. Again, there is currently a limit of approximately 2 billion sequences per individual SRA BLAST query.

Direct downloading of fasta and fastq format
The SRA provides a tool that can be used to download data directly in fasta or fastq format. You must provide one or more SRA Experiment (SRX, DRX, or ERX) accessions in a comma-separated list. The same filtering inputs available in the Run Browser (described above) are available here to restrict the number of returned reads. Certain reads can also be clipped to remove low quality data from the download. If more than one Run accession in the list is checked, all data will be downloaded into a single fasta or fastq file, rather than per-accession files. Note that the output format of this tool is pre-defined and cannot be adjusted at the time of download. Users with specific formatting needs (e.g., for downstream analysis) are encouraged to use the SRA Toolkit to download and convert the data (described above).

Downloading metadata associated with SRA data files
SRA data files do not contain any information about the metadata (sample information, etc.) linked to the data themselves. The SRA provides a few tools to allow downloading of metadata in batch. Note that these tools differ from the Entrez Experiment, BioSample, and BioProject reports for a given dataset and may not contain all relevant metadata.
Viewing and downloading tabular metadata with the SRA Run Selector
The SRA Run Selector can be used to view metadata from one or more projects (SRA Study accessions – SRP, DRP, or ERP) entered into the field at the top of the page. The Run Selector provides a table view of library preparation and sample attribute metadata. The table can be filtered by sample attribute(s), accessions, etc. The “Get Metadata” button can be used to download a table (.txt, tab-delimited) of all or selected metadata.

Command line access to metadata with the SRA Run Info CGI
Users can access the SRA Run Info CGI either through a browser or using a command line tool like wget.
wget -O <file_name.csv> 'http://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?save=efetch&db=sra&rettype=runinfo&term=<query>'
As a parallel to the above example in the Run Selector,
wget -O ./SRP001599_info.csv 'http://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?save=efetch&db=sra&rettype=runinfo&term= SRP001599'
Will return essentially the same information. Note that the CGI returns data in a comma-separated value (.csv) format, rather than the tab-delimited format of the Run Selector. The last component, <query>, can contain any set of Entrez parameters. Users may refine a search using Entrez and then copy over the search terms to a script for batch downloading. As an example, the search string
"Homo sapiens"[Organism] AND "cancer"[All Fields] AND "cluster_public"[prop] AND "strategy wgs"[Properties]
Will return these results in an Entrez search of the SRA. The equivalent Run Info CGI search would be
wget -O ./query_results.csv 'http://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?save=efetch&db=sra&rettype=runinfo&term="Homo sapiens"[Organism] AND "cancer"[All Fields] AND "cluster_public"[prop] AND "strategy wgs"[Properties]'
Note that Entrez groups by Experiment accession, but that the CGI does not. It is, therefore, to be expected that the Run Info CGI will return a longer list of results than Entrez, but will still contain the same datasets.
- Download Guide - SRA HandbookDownload Guide - SRA Handbook
Your browsing activity is empty.
Activity recording is turned off.
See more...