

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

Previous attempts to develop compounds that are highly selective for M1 or other specific muscarinic acetylcholine receptor (mAChR) subtypes have failed because of the high conservation of the ACh binding site and difficulty in developing truly specific compounds. Basic and clinical studies suggest that mAChR activation has the potential to be disease modifying, as well as to provide palliative cognitive therapy. Probes developed from these efforts will greatly advance the current state of the art by aiding in the understanding of M1's role in cell-based physiology and may extend the clinical understanding of psychotic and cognitive symptoms associated with neurodegenerative disorders such as Alzheimer's Disease and schizophrenia. This probe report describes ML071 (CID-25010775), a highly selective, potent M1 agonist that activates M1 by virtue of binding at an allosteric site in the third extracellular loop of the M1 receptor. The compound represents a "best in class" probe that provides unprecedented mAChR selectivity, clean ancillary pharmacology, saline solubility, and central penetrance. Furthermore, it can be employed for in vitro and in vivo studies to examine the role of selective M1 receptor activation.
Assigned Assay Grant #: X01 MH077606-01
Screening Center Name & PI: Vanderbilt Screening Center for GPCRs, Ion Channels, and Transporters, David Weaver
Chemistry Center Name & PI: Vanderbilt Specialized Chemistry Center for Accelerated Probe Development, Craig Lindsley
Assay Submitter & Institution: P. Jeffrey Conn, Vanderbilt University
PubChem Summary Bioassay Identifier (AID): AID-1798
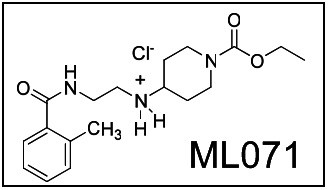
Probe Structure & Characteristics
Ethyl 4-(2-methylbenzamido)ethylamino)piperidine-1-carboxylate hydrochloride
| CID | Target Name | IC50/EC50 (nM) [SID, AID] | Anti-target Name(s) | IC50/EC50 (μM) [SID, AID] | Selectivity | Secondary Assay(s) Name: IC50/EC50 |
|---|---|---|---|---|---|---|
| 25010775 (ML071) | M1 |
198
[SID-56353039, AID-626, AID-1488, AID-1741, AID-1744] | M2–M5 | >30
μM [SID-56353039, AID-626, AID-1488, AID-1741, AID-1744, AID-1470, AID-1767, AID-1764, AID-1757, AID-1508, AID-1788] | >263-fold | M1 Y381A 379 nM [SID-56353039, AID-1743] |
Recommendations for the scientific use of this probe
This probe can be used for both in vitro and in vivo to study the role of selective M1 receptor activation. The compound possesses unprecedented selectivity versus M2–M5 and against a large panel of GPCRs, ion channels and transporters. Moreover, the probe is centrally penetrant and soluble in saline (>25 mg/mL).
Specific Aim
To identify small molecule agonists of M1 muscarinic receptor that are cell permeable, exhibit submicromolar potency, and show greater than 10 fold selectivity over other muscarinic family members M2, M3, M4 and M5. This probe report describes a potent, highly selective M1 allosteric agonist (CID 25010775 (ML071), SID-56353039) that activates M1 by virtue of binding at an allosteric site in the third extracellular loop of the M1 receptor. The probe is ‘best in class’ providing unprecedented mAChR selectivity, clean ancillary pharmacology, soluble in saline (~25 mg/mL) and centrally penetrant.
Significance
Previous attempts to develop compounds that are highly selective for M1 or other specific mAChR subtypes have failed because of the high conservation of the Ach binding site and difficulty in developing truly specific compounds (Bonner et al. 1997, 1998; Felder et al. 2000; Bymaster et al. 2003). The lack of highly selective compounds has made it impossible to definitively determine the behavioral and clinical effects of these receptors. In numerous Phase II and III clinical trails, pan-mAChR agonists were shown to improve cognitive performance in AD patients, but the GI-and/or cardiovascular side effects, resulting from activation of peripheral mAChRs, were deemed intolerable and the trials were discontinued (Eglen et al. 2001; Tarsy et al. 2006). Importantly, several pan-mAChR agonists demonstrated decline of Aβ42 in the cerebral spinal fluid of AD patients, suggesting that mAChR activation has the potential to be disease modifying as well as providing palliative cognitive therapy (Bodick et al. 1997). More recent studies in 3xTg-AD mice further support a disease modifying role for mAChR activation, and several Ph III trials demonstrated that mAChR activation lowered Aβ42 in patients (Caccamo et al. 2006). Interestingly, the M1/M4 preferring xanomeline, in addition to improving cognitive performance, had robust therapeutic effects on the psychotic symptoms and behavioral disturbances associated with AD and recently published clinical trial data indicates efficacy in schizophrenic patients (Bodick et al. 1997; Shekhar et al. 2008). Probes developed from these efforts will greatly advance the current state of the art by aiding in the understanding of M1’s role in cell-based physiology and may extend the clinical understanding of psychotic and cognitive symptoms associated with neurodegenerative disorders like Alzheimer’s Disease and schizophrenia.
Rationale
In recent years, major advances have been made in the discovery of highly selective agonists of other G protein-coupled receptors (GPCRs) that act at an allosteric site rather than the orthosteric binding site (May et al. 2003). By screening for compounds that act at an allosteric site on the receptor, it is anticipated that compounds that selectively activate M1 versus the other muscarinic subtypes may be identified. While allosteric M1 agonists have been identified, AC-42 and TBPB, they both suffer from undesirable ancillary pharmacology, poor physicochemical properties, poor pharmacokinetics and/or limited CNS penetration (Spalding et al. 2002; Jones et al. 2008). Thus, to truly enable the biomedical community to dissect the relative contributions of selective M1 activation in preclinical models of AD and schizophrenia and to understand the role of M1 in the pronounced efficacy of the M1/M4 preferring xanomeline, improved M1 probes are required.
Screening center information
Assay Implementation and Screening
PubChem Bioassay Name: Discovery of novel allosteric modulators of the M1 muscarinic receptor: Agonist
List of PubChem bioassay identifiers generated for this screening project (AIDS): AID-626, AID-1488, AID-1741, AID-1508, AID-1470, AID-1744, AID-1767, AID-1764, AID-1757, AID-1743, AID-1788.
PubChem Primary Assay Description
- Hamster Ovary (CHO) cells containing M1 receptor (ATCC#CRL-1985) were plated at 10,000 cells/well in assay media (F12 (Ham), 10% FBS, 2 millimolar GlutaMAX (Invitrogen), 20mM HEPES) in 384 well plates.
- The plates were incubated overnight at 37 degrees C in 5% CO2.
- Media was removed and assay buffer (Hanks Balanced Salt Solution, 20 millimolar HEPES, 2.5 millimolar Probenecid, pH 7.4) containing 4.0 micromolar Fluo4-AM dye (Invitrogen) was added.
- Cells were incubated for 45 minutes (37 degrees C, 5% CO2) for dye loading.
- Cell plates were loaded into the Hamamatsu FDSS equipped with 480 nanometer excitation and 540 nanometer emission filters.
- 10 micromolar test compound in assay buffer + 0.1% DMSO was added at 5 seconds; simultaneously, the plate was kinetically imaged.
- Subsequently, 8 nanomolar acetylcholine (EC80) in assay buffer was added at 197 seconds and imaging continued for a total of 4 minutes acquisition time.
- 0.1% DMSO, compound vehicle, and 80nM acetylcholine (ECMAX) were added to each plate as controls.
- Compounds that stimulated an increase in intracellular calcium upon their addition deviating by more than three standard deviations from the test populations were identified as “hits”.
Summary of Screen
For the discovery of novel allosteric agonists of the M1 muscarinic receptor, we completed the primary screen using a real-time cell-based assay against the full 65K library. The assay performed very well (Z′ averaged 0.7) and we identified approximately 2000 putative agonist primary hits. The compounds clustered nicely into different structural classes with multiple representative analogs in each cluster. The agonist compounds were chosen as the focus of this probe effort, but we previously filed a probe report for the first truly selective small molecule M1 antagonist, SID-56373925.
For the confirmation screen, 1666 agonist hits were reordered from Biofocus-DPI. The compounds were tested in duplicate against M1/CHO cells, and in parallel, counter screened against M4/Gqi5 CHO cells. Compounds showing initial selectivity for M1 over M4 were tested in triplicate in a 10 point dose-response series against the muscarinic panel (M1–M5). Compounds showing selectivity for M1 were then tested as a concentration series in the presence of atropine. Pubchem CID 644390 and CID 647412 were selective (>50-fold) for M1 vs. M4 with M1 EC50s from the DPI DMSO stock of ~ 1 μM (Figure 1) and sensitive to block by atropine.

Figure 1
M1 vs. M4 selective Agonist Leads from the HTS.
We then resynthesized the two HTS leads to confirm structure and activity (Scheme 1). In the event, commercial mono- N-Boc diaminoethane was acylated with either 2-chlorobenzoyl chloride (2) or 2-thienyl acyl chloride (3) to deliver 4 and 5, respectively. The Boc group was removed by exposure to 4N HCl/dioxane to provide the corresponding HCl salts 6 and 7. Finally, a reductive amination sequence employing ethyl-4-oxopiperidine-1-carboxylate (8) with a polymer-supported hydride delivered the original HTS leads, CID 644390 and CID 647412 in overall yields for the three step sequence in excess of 70%.

Scheme 1
Resynthesis of M1 vs. M4 selective Agonist Leads from the HTS; 70% overall yield.
The resynthesized CID 644390 and CID 647412 confirmed, with M1 EC50s of 804 nM and 1.74 μM, respectively. Importantly, both compounds proved to be highly selective versus M2–M5, affording no activation at concentrations exceeding 50 μM (Figure 2). Based on this unprecedented mAChR selectivity, we assumed these ligands must be binding at an allosteric sitetoelicit receptor activation. Competition binding studies with the orthosteric antagonist [3H]-NMS demonstrated that CID 644390 (SID-842134) and CID 647412 (SID-845074) displace the radioligand only at very high concentrations, suggesting they do in fact bind at an allosteric site (Figure 3). However, these initial hits do not meet MLPCN probe criteria, so we initiated a chemical lead optimization campaign to develop a selective M1 allosteric agonist that meets MLPCN criteria.

Figure 2
In vitro pharmacological profile of resynthesized CID 644390 and CID 647412 on M1–M5.
![Figure 3. Competition Binding with [3H]-NMS.](/books/NBK47340/bin/ml071f3.gif)
Figure 3
Competition Binding with [3H]-NMS.
Probe Chemical Lead Optimization Strategy
We employed an iterative parallel synthesis approach for the optimization of CID 644390 and CID 647412, as the scaffolds were modular and readily amenable to this approach. We simultaneously investigated three areas of the scaffold (Figure 4) employing the synthetic route depicted in Scheme 1, and quickly noticed (Table 1), that responsive SAR was only observed for the amide moiety (blue). No modifications to the linker region (yellow) were tolerated, and this included substitutions along the ethyl chain, capping either terminal nitrogen with a methyl, or homologation of the linker. Similar flat SAR was observed for the ethyl carbamate moiety (green). No alternatives (amides, alkyl/phenyl or differen carbamates) were tolerated. Of 40 analogs prepared, only six (15%) displayed any measurable M1 activity. However, the six active compounds all possessed sub-micromolar EC50s at M1 (152 nM to 459 nM), and all were completely selective versus M2–M5 (EC50s >50 μM). Thus, all six met the minimum criteria for an MLPCN probe. We then counter-screened the six compounds versus D2 as a mean to distinguish the probe candidates, as activity at D2 would greatly diminish the value of an M1 probe to explore the role of selective M1 activation in modulating the pathophysiology of schizophrenia. This counter-screen eliminated four potential probe compounds (low micromolar D2 IC50s) and left CID 650889 and CID 25010776 as contenders (D2 IC50s >10 μM). The two potential probes were then resynthesized and screened as the corresponding HCl salts 650889 (25010774) and 25010775 (ML071). After completing n=3 CRCs for the potential probes, the M1 EC50s for CID 25010774 and CID 25010775 (ML071) were 152±8.4 nM (85.1±3.3% ACh Max) and 198±13.2 nM (80.52±7.6% ACh Max) with complete selectivity versus M2–M5 (Figure 5a). To confirm that these compounds are indeed allosteric agonists, we evaluated them on a Y381A mutant M1 cell line. Allosteric agonists of M1 can be differentiated from orthosteric agonists by their ability to activate the receptor in which there is a single point mutation (Y381A) in the orthosteric binding site that renders the receptor insensitive to acetylcholine or orthosteric agonists. As shown in Figure 5b, the Y381A mutation causes three order of magnitude right-shift in the ACh CRC and TBPB, the prototypical M1 allosteric agonist, retains some efficacy on this line. In contrast, the initial HTS leads (CID 644390 and CID 647412) and the two probe candidates (CID 25010774 and CID 25010775 (ML071)) remain fully efficacious on this mutant line. In fact, the EC50s for CID 650889 and CID 25010776 shift less than 2-fold (Y381A M1 EC50s = 304±56 nM and 379±91 nM, respectively). These data, coupled with competition binding studies with [3H]-NMS (Figure 6), clearly indicate that these new M1 agonists activate the receptor through binding at an allosteric site.
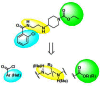
Figure 4
Chemical Optimization Plan for CID 644390 and CID 647412.
Table 1
Structure-Activity Relationships for Analogs of CID 644390 and CID 647412.
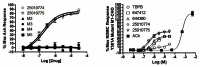
Figure 5
A) Full CRCs for CID 25010774 and CID 25010775 for wt M1–M5; B) CRCs for TBPB, ACh, the initial HTS leads CID 644390 and CID 647412 and the probe candidates CID 25010774 and CID 25010775.
![Figure 6. Competition [3H]-NMS binding for the initial HTS leads CID 644390 and CID 647412 and the probe candidates CID 25010774 and CID 25010775 relative to the orthosteric antagonist atropine.](/books/NBK47340/bin/ml071f6.gif)
Figure 6
Competition [3H]-NMS binding for the initial HTS leads CID 644390 and CID 647412 and the probe candidates CID 25010774 and CID 25010775 relative to the orthosteric antagonist atropine.
A major liability with TBPB, the leading allosteric M1 agonist in the literature to date, is the fact that TBPB is a pan-mAChR antagonist at higher concentrations at M2–M5. This undesired ancillary pharmacology greatly limits the utility of TBPB to cleanly dissect the role of selective M1 activation in vitro and in vivo. Gratifyingly (Figure 7), the initial HTS leads (CID 644390 and CID 647412) and the two probe candidates (CID 25010774 and CID 25010776) show no significant antagonism of M2–M5 and further distinguish themselves as superior M1 allosteric agonist probes.

Figure 7
Functional antagonism comparison of TBPB and for the initial HTS leads CID 644390 and CID 647412 and the probe candidates CID 25010774 and CID 25010775.
In parallel, to further confirm that the two probe candidates (CID 25010774 and CID 25010775 (ML071)) are indeed activating M1 at an allosteric site, we evaluated both probe candidates against a mutant line we had in house targeting the third extracellular loop (e3) that is far removed from the orthosteric binding site. A (K392D/E397V/E410H) triple mutant abolishes the ability of the two probe candidates (CID 25010774 and CID 25010775 (ML071)) to activate M1 (Figure 8). Evaluation of the three individual single point mutants demonstrated that the E401H is critical for M1 activation with the two probe candidates (CID 25010774 and CID 25010775 (ML071)), suggesting they bind to the e3 loop of M1, far removed from the orthosteric binding site.
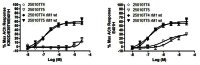
Figure 8
Further data with an M1 triple mutant (K392D/E397V/E401H) for the probe candidates CID 25010774 and CID 25010775, and identification of a lone single point mutant (E401H) that diminishes M1 activation.
At this point, the Lead Profiling Screen (68 GPCRs, ion channels and transporters) from MDS Pharma was performed on the two probe candidates (CID 25010774 and CID 25010776) to attempt to distinguish which would be promoted to probe status. CID 25010775 (ML071) (SID-56353039) possessed superior ancillary profile to both TBPB and CID 25010774 (Figure 9), with only three activities >50% at 10 μM, and was thus declared an MLPCN probe. CID 25010775 (ML071) has the following IUPAC nomenclature: ethyl 4-(2-methylbenzamido)ethylamino) piperidine-1-carboxylate.
Figure 9
MDS Pharma Lead Profiling Screen of 68 GPCRs, ion channels and transporters against CID 25010775 (SID-56353039) at a concentration of 10 μM.

CID 25010776 A Best-in-Class M1 allosteric agonist probe
Finally, a brain/plasma study was conducted to determine if our M1 allosteric agonist probe (CID 25010775 (ML071)) was centrally penetrant after systemic dosing, as this would add significant value to the probe for the biomedical research community. When converted to the mono- HCl salt, CID 25010775 (ML071) displayed excellent solubility across pharmaceutically acceptable vehicles, as well as DMSO (> 100 mM) and water (homogeneous solutions up to 25 mg/mL). For the brain/plasma study, CID 25010775 (ML071) (SID-56353039) as the mono-HCl salt, was dosed in water at 10 mg/kg i.p. to Sprauge-Dawley rats (Figure 10). A brain/plasma ratio of 4.2 was observed, with the compound preferentially portioning into the brain. This is an excellent profile for a CNS agent, with brain levels in the μM range for up to 5 hours post dose - >8-fold above the EC50 for M1 activation. Importantly, the animals were closely monitored, and were healthy, with no signs of classical pan-mAChR activation (SLUD – salivation, lacrimation, urination and defecation) indicating that the in vitro mAChR selectivity profile was mirrored in vivo. Thus, CID 25010775 (ML071) has utility as both an in vitro and in vivo probe for selective M1 activation by an allosteric mechanism.
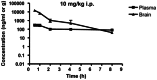
Figure 10
Brain/plasma study with SID-56353039.
Scheme 2 below highlights the optimized route to prepare CID 25010776 as the mono-HCl salt CID 25010775 (ML071) in 82.5% overall yield. All of the reagents are commercially available from Aldrich Chemical company. MLS#s: MLS002279948, MLS002279949, MLS002279950, MLS002279951, MLS002279952, MLS002279953
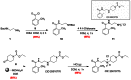
Scheme 2
Optimized synthesis of CID 25010776 as free base and mono-HCl salt, 25010775: overall yield of 82.5%.
Bibliography
- Bodick NC, Ofen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, Shannon HE, Tollefson GD, Rasmussen K, Bymster FP, Hurley DJ, Potter WZ, Paul SM. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer’s disease. Arch. Neurol. 1997;54:465–473. [PubMed: 9109749]
- Bonner TI, Buckley NJ, Young AC, Brann MR. Identification of a family of muscarinic acetylcholine receptor gene. Science. 1987237:527–532. [PubMed: 3037705]
- Bonner TI, Young AC, Buckley NJ, Brann MR. Cloning and expression of the human and rat m5 muscarinic receptor gene. Neuron. 1988;1:403–410. [PubMed: 3272174]
- Bymaster FP, McKinzie DL, Felder CC, Wess J. Use of M1–M5 muscarinic receptor knockout mice as novel tools to delineate the physiological roles of the muscarinic cholinergic system. Neurochem. Res. 2003;28:437–442. [PubMed: 12675128]
- Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, LaFerla FM. M1 receptors play a cnetral role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49:671–682. [PubMed: 16504943]
- Eglen RM, Choppin A, Dillon MP, Hedge S. Muscarinic receptor ligands and their therapeutic potential. Curr. Opin. Chem. Biol. 1999;3:426–432. [PubMed: 10419852]
- Felder CC, Bymaster FP, Ward J, DeLapp N. Therapeutic opportunities for muscarinic receptors in the central nervous system. J. Med. Chem. 2000;43:4333–4353. [PubMed: 11087557]
- Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane A, Bridges TM, Kennedy JP, Bradley SR, Peterson T, Baldwin RM, Kessler R, Deutch A, Lah JL, Levey AI, Lindsley CW, Conn PJ. Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor reduces amyloid processing and produces antipsychotic-like activity in rats. J. Neurosci. 2008;28(41):10422–10433. [PMC free article: PMC2577155] [PubMed: 18842902]
- May LT, Christopoulos A. Allosteric modulators of G-protein–coupled receptors. Curr Opin Pharmacol. 2003;3(5):551–6. [PubMed: 14559102]
- Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am. J. Psychiatry. 2008;165:1033–1039. [PubMed: 18593778]
- Spalding TA, Trotter C, Skajaerbaek N, Messier TL, Currier EA, Burstein ES, Li D, Hacksell U, Brann MR. Discovery of an ectopic site activation site on the M1 muscarinic receptor. Mol. Pharm. 2002;61:1297–1302. [PubMed: 12021390]
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- Review Discovery and development of the a highly selective M(1) Positive Allosteric Modulator (PAM).[Probe Reports from the NIH Mol...]Review Discovery and development of the a highly selective M(1) Positive Allosteric Modulator (PAM).Bridges TM, Lewis LM, Dawson ES, Weaver CD, Lindsley CW. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Discovery and development of a second highly selective M(1) Positive Allosteric Modulator (PAM).[Probe Reports from the NIH Mol...]Review Discovery and development of a second highly selective M(1) Positive Allosteric Modulator (PAM).Bridges TM, Reid PR, Lewis LM, Dawson ES, Weaver CD, Wood MR, Lindsley CW. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Discovery of a Highly Selective in vitro and in vivo M4 Positive Allosteric Modulator (PAM).[Probe Reports from the NIH Mol...]Review Discovery of a Highly Selective in vitro and in vivo M4 Positive Allosteric Modulator (PAM).Lewis LM, Bridges TM, Niswender CM, Weaver CD, Lindsley CW. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Discovery of the first mAChR 5 (M5) selective ligand, an M5 Positive Allosteric Modulator (PAM).[Probe Reports from the NIH Mol...]Review Discovery of the first mAChR 5 (M5) selective ligand, an M5 Positive Allosteric Modulator (PAM).Bridges TM, Lewis LM, Weaver CD, Lindsley CW. Probe Reports from the NIH Molecular Libraries Program. 2010
- Pharmacological characterization of LY593093, an M1 muscarinic acetylcholine receptor-selective partial orthosteric agonist.[J Pharmacol Exp Ther. 2011]Pharmacological characterization of LY593093, an M1 muscarinic acetylcholine receptor-selective partial orthosteric agonist.Watt ML, Schober DA, Hitchcock S, Liu B, Chesterfield AK, McKinzie D, Felder CC. J Pharmacol Exp Ther. 2011 Aug; 338(2):622-32. Epub 2011 May 10.
- Discovery of a Highly Selective in vitro and in vivo M1 Allosteric Agonist Probe...Discovery of a Highly Selective in vitro and in vivo M1 Allosteric Agonist Probe - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...