

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

Defective bone mineralization (osteomalacia) occurs in tissue non-specific alkaline phosphatase (TNAP) deficiency (hypophosphatasia). TNAP functions to maintain normal bone mineralization via hydrolysis of inorganic pyrophosphate (PPi), a bone mineralization inhibitor. This study sought to examine the role of PHOSPHO1, a soluble phosphatase that has specificity for phosphoethanolamine and phosphocholine, in increasing the inorganic phosphate (Pi)/inorganic pyrophosphate (PPi) ratio inside matrix vesicles (MVs) and, thus, controlling the first step of initiation of hydroxyapatite crystal deposition inside MVs. The identified molecular probe ML086 (CID-1674999) is a biochemical inhibitor of PHOSPHO1. PHOSPHO1-specific inhibitors may serve as novel tools to examine the functional role of this enzyme in skeletal mineralization and soft tissue ossification abnormalities, as well as to help elucidate its mechanism of action in diseases such as osteoarthritis, osteoporosis, and arterial calcification.
Assigned Assay Grant #: 1 R03 MH084086-01
Screening Center Name & PI: Conrad Prebys Center for Chemical Genomics (formerly Burnham Center for Chemical Genomics) & Dr. John C. Reed
Chemistry Center Name & PI: Conrad Prebys Center for Chemical Genomics (formerly Burnham Center for Chemical Genomics) & Dr. John C. Reed
Assay Submitter & Institution: Dr José Luis Millán & Sanford-Burnham Medical Research Institute (formerly Burnham Institute for Medical Research)
PubChem Summary Bioassay Identifier (AID): AID-1574
Probe Structure & Characteristics
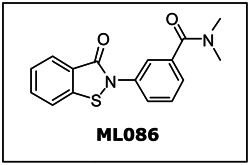
| CID/ML | Target Name | IC50/EC50 (nM) [SID, AID] | Anti- target Name(s) | IC50/EC50 (μM) [SID, AID] | Selectivity | Secondary Assay(s) Name: IC50/EC50 (nM) [SID, AID] |
|---|---|---|---|---|---|---|
| CID-16749996 ML086 | PHOSPHO1 | 139 nM IC50 SID-57287582 AID-1666 | TNAP | >100 μM IC50
SID-57287582 AID-1056 | >719 | PMM2: >100,000 nM IC50 SID-57287582 AID-1655 PMI: >50,000 nM IC50 SID-57287582 AID-1535 |
Recommendations for the scientific use of this probe
A newly identified soluble phosphatase, PHOSPHO1, with specificity for phosphoethanolamine (PEA) and phosphoserine (PS) present in matrix vesicles (MVs), is responsible for increasing the local concentration of Pi inside MVs to change the Pi/PPi ratio to favor precipitation of hydroxyapatite (HA) seed crystals. As tissue non-specific alkaline phosphatase (TNAP) is also in the same biological compartment, PHOSPHO1-specific inhibitors will enable us to probe and clarify the functional involvement of this enzyme in skeletal mineralization and soft tissue ossification abnormalities. This will help to elucidate the mechanism of action for diseases such as osteoarthritis, osteoporosis, and arterial calcification.
1. Scientific Rationale for Project
Specific Aims
The main aim of this project was to screen large comprehensive chemical libraries to identify lead compounds for PHOSPHO1-specific inhibitors that will enable the elucidation of the functional involvement of this enzyme in skeletal mineralization and related biological phenomena.
Background and Significance
During the process of endochondral bone formation, osteoblasts mineralize the extracellular matrix (ECM) by promoting the initial formation of crystalline hydroxyapatite in the sheltered interior of membrane-limited matrix vesicles (MVs) and by modulating matrix composition to further promote propagation of apatite outside of the MVs. All available data indicate that controlled bone mineralization depends on a regulated balance of the following factors: the concentrations of Ca2+ and inorganic phosphate (Pi), the presence of fibrilar collagens (e.g., type I in bone; Types II and X in cartilage) and the presence of adequate concentrations of mineralization inhibitors, i.e., inorganic pyrophosphate (PPi), and osteopontin (OPN). Three molecules present in osteoblasts have so far been identified as affecting the controlled deposition of bone mineral by regulating the extracellular levels of PPi, and in turn of OPN, i.e., tissue-nonspecific alkaline phosphatase (TNAP); NPP1 (a nucleotide pyrophosphatase/phosphodiesteraseisozyme) and the ANK gene product. A deficiency in the TNAP isozyme causes the inborn-error-of-metabolism known as hypophosphatasia and the study of this disease has provided the best evidence of the importance of TNAP for bone mineralization. TNAP is the only tissue-nonrestrictedisozyme of a family of four homologous human alkaline phosphatase (AP) genes (EC. 3.1.3.1) (1) that additionally includes the placental (PLAP), germ cell (GCAP) and intestinal (IAP) AP genes. Expressed as an ecto-enzyme anchored via a phosphatidylinositolglycan moiety, TNAP has been demonstrated to play an essential physiological role during bone matrix mineralization. Specifically, defective bone mineralization (osteomalacia) occurs in TNAP deficiency (hypophosphatasia) (2). The severity of hypophosphatasia is variable and modulated by the nature of the TNAP mutation (3–6). Unlike most types of rickets or osteomalacia neither calcium nor inorganic phosphate levels in serum are subnormal in hypophosphatasia. In fact hypercalcemia and hyperphosphatemia may exist and hypercalciuria is common in infantile hypophosphatasia (7). The clinical severity in hypophosphatasia patients varies widely. The different syndromes, listed from the most severe to the mildest forms, are: perinatalhypophosphatasia, infantile hypophosphatasia, childhood hypophosphatasia, adult hypophosphatasia, odontohypophosphatasia and pseudohypophosphatasia (7). These phenotypes range from complete absence of bone mineralization and stillbirth to spontaneous fractures and loss of decidual teeth in adult life. Inactivation of the mouse TNAP gene (Akp2) phenocopies the infantile form of human hypophosphatasia (8, 9). In bone, TNAP is confined to the cell surface of osteoblasts and chondrocytes, including the membranes of their shed MVs (10, 11). In fact, by an unknown mechanism, MVs are markedly enriched in TNAP compared to both whole cells and the plasma membrane (12). It has been proposed that the role of TNAP in the bone matrix is to generate the inorganic phosphate needed for hydroxyapatite crystallization (13–15). However, TNAP has also been hypothesized to hydrolyze the mineralization inhibitor PPi (16) to facilitate mineral precipitation and growth (17–19). Electron microscopy revealed that TNAP-deficient MVs, in both humans and mice, contain apatite crystals, but that extravesicular crystal propagation is retarded (20). This growth retardation could be due to either the lack of TNAP'spyrophosphatase function or the lack of inorganic phosphate-generation. Our recent studies have provided compelling proof that the function of TNAP in bone tissue consists of hydrolyzing PPi to maintain a proper concentration of this mineralization inhibitor to ensure normal bone mineralization. PPi is primarily generated by the members of the nucleotide pyrophosphatase/phosphodiesterase (NPP) family of isozymes. Plasma cell membrane glycoprotein-1 (PC-1; more correctly termed NPP1) is plasma membrane-bound, whereas autotaxin (NPP2) is secreted and B10 (NPP3) is abundant in intracellular spaces (21). All three isozymes are expressed in a wide variety of tissues, including bone and cartilage (22), and they all have the common ability to hydrolyze diesters of phosphoric acid into phosphomonoesters. NPPs have been implicated in various processes, including bone mineralization, signaling by insulin and by nucleotides, and the differentiation and motility of cells (23). However, NPPs are known primarily as suppliers of intra- and extracellular PPi (24). Similar to skeletal TNAP expression, NPP1 is highly abundant on the surfaces of osteoblasts and chondrocytes as well as on the membrane of their MVs (25, 26). NPP1 has a role in inhibiting hydroxyapatite precipitation by its PPi-generating property. This proposed function has been supported by in vitro studies where cells transfected with the NPP1 cDNA resulted in elevated levels of PPi in osteoblast-derived MVs, accompanied by decreased matrix mineralization (27). It is also thought that glycoprotein-1 participates in osteblastic mineralization (28). Moreover, Enpp1−/− mice develop hypermineralization abnormalities such as ossification of the posterior longitudinal ligament of the spine, diffuse idiopathic skeletal hyperostosis, ankylosing spinal hyperostosis and pathological soft-tissue ossification, including arterial calcification (29, 30). Conversely, while NPP1 deficiency results in hypermineralization, elevated expression of NPP1 is associated with MV-mediated calcium pyrophosphate dihydrate (CPPD) matrix calcification in the knee meniscal cartilage during aging (31, 32). Interestingly, as in NPP1 overexpressing tissues, abnormal CPPD precipitation has also been observed in association with TNAP deficiency. While the excess of PPi in hypophosphatasia patients is primarily linked with rickets and osteomalacia, subjects with the disease may also develop pathological CPPD mineralization of the articular cartilage (33).
Similar to NPP1, the ankylosis protein (ANK) has a role in mineralization by contributing to the extracellular supply of PPi. However, unlike NPP1, ANK appears to function as a transmembrane PPi-channeling protein, allowing PPi molecules to passage through the plasma membrane from the cytoplasm to the outside of the cell (34). ANK protein is detectable in many tissues, yet its expression is particularly strong in the cartilage of joints (34). Cell surfaces of osteoblasts and chondrocytes appear to be abundant in ANK protein (36), but in contrast to NPP1 and TNAP, it is not present in the membranes of MVs. ANK was identified in a naturally occurring mutant mouse strain that had characteristics of progressive ankylosis, thus the designation ank/ankmice (35). These animals develop hydroxyapatite crystals in articular surfaces and synovial fluids. As Enpp1−/− mice, ank/ankmice display pathological abnormalities that mimic several arthritic diseases, including ectopic calcification, cartilage erosion and osteophyte formation seen in osteoarthritis, and vertebral fusion observed in ankylosisspondylitis patients (37–40).
It has also been shown that normalizing calcium and Pi concentrations corrects the hyperosteoidosis of rickets patients (41). Since extracellular calcium is more tightly regulated than extracellular Pi concentrations, Pi may be the critical element in the induction of mineral crystals in a given ECM. The production by osteoblasts of an inhibitor of ECM mineralization like PPi is counter-intuitive. It suggests a model whereby the removal of an inhibitor rather than the synthesis of an inducer of mineralization would explain why ECM mineralization occurs in bone. Consistent with this model we have shown that removal of PPi via TNAP action and the presence of a fibrilar collagen-rich scaffold are two conditions necessary to induce mineralization of bone or any ECM. Our data also indicate that the Pi/PPi ratio is of fundamental significance for bone ECM mineralization. This is in agreement with the observation that Akp2− / −mice that have abnormally high extracellular PPi levels have hyperosteoidosis, while [Akp2− / −; Enpp1− / −] and [Akp2− / −; ank/ank] mice have normal mineralization of the skull and normal extracellular PPi concentration (42,43). Thus, in the bone ECM, while the extracellular Pi concentration is fairly constant, TNAP’s enzymatic degradation of PPi controls the Pi/PPi ratio to favor crystallization of HA outside the MVs along collagen fibrils. But, why are Akp2−/− mice born with a mineralized skeleton and still contain HA crystals inside their MVs? As TNAP sits on the outer surface of the MV membrane there is no TNAP-mediated hydrolysis of PPi inside the MVs. Therefore, it is likely that another enzyme is responsible for either cleaving PPi or elevating the intravesicular concentration of Pi so as to achieve a Pi/PPi ratio conducive for crystallization. We proposed and tested our hypothesis in this research that PHOSPHO1, a soluble phosphatase (44, 45) that has specificity for phosphoethanolamine and phosphocholine (46), plays the important role of increasing the Pi/PPi ratio inside MVs and thus controls the first step of initiation of HA crystal deposition inside MVs.
2. Project Description
a. The original goal for probe characteristics
The original goal was to find compounds that had minimally less that 1 µM potency, but were selective against tissue specific alkaline phosphatase. None of the existing three inhibitors (SCH-20676, lansoprazole, ebselen) from the original CPDP met the 1 µM potency barrier.
b. Assay implementation and screening
i. PubChem Bioassay Name(s), AID(s), Assay-Type (Primary, DR, Counterscreen, Secondary)
| PubChemBioAssay Name | AIDs | Probe Type | Assay Type | Assay Format | Assay Detection & wellformat |
|---|---|---|---|---|---|
| uHTS absorbance assay for the identification of compounds that inhibit PHOSPHO1. | 1565 | Inhibitor | Primary | biochemical | Absorbance (@620 nm, BIOMOL)1536 |
| SAR assay for compounds that inhibit PHOSPHO1 | 1666 | Inhibitor | SAR | biochemical | Absorbance at 630 nm, 384 |
| Anti-target in Vitro TNAP Dose Response Luminescent Assay for SAR Study | 1056 | Inhibitor | Anti-target Counterscn | biochemical | Luminescence, 384 |
| Counter screen SAR assay for PMM2 inhibitors via a fluorescence intensity assay | 1655 | Inhibitor | Secondary Assay for specificity | biochemical | Fluorescence 384 |
| Confirmation of compounds inhibiting phosphomannoseisomerase (PMI) via a fluorescence intensity assay | 1535 | Inhibitor | Secondary Assay for specificity | biochemical | Fluorescence 384 |
ii. Assay Rationale & Description
This biochemical assay employs a colorimetric readout based on the enzyme's ability to liberate phosphate from phosphoethanolamine and its reaction with the Biomol Green reagent. The primary screening protocol is described below.
Assay materials
- PHOSPHO1 was obtained from the assay provider's laboratory. The construct was designed to express PHOSPHO1 fused to a V5 epitope and 6 His-tag at the C-terminus.
- Assay Buffer: 20mM MES-NaOH pH 6.7, 2mM MgCl2, 0.0125% Tween-20, 0.01% BSA.
Table 1Reagents used for the uHTS experiments
| Reagent | Vendor |
|---|---|
| Recombinant human PHOSPHO1 produced in E. coli from an arabinose inducible obtained expression clone | Invitrogen-- pBAD TOPO TA vector from a cDNA corresponding to Met19-Cys267 of human PHOSPHO1 |
| BIOMOL Green (AK-111) | BIOMOL |
| SCH-202676: previously identified as an inhibitor of PHOSPHO1 as positive control | Calbiochem, Cat No. 565645 |
The following uHTS protocol was developed
- 1.5ul of assay buffer (20mM MES-NaOH, 2mM MgCl2, 0.0125% Tween 20, 0.01% BSA) are added to each well in columns 1 and 2 of a black/clear bottom Corning assay plate (cat # 3891).
- 1.5ul of assay buffer containing 2.5ng/ul PHOSPHO1 are added to the wells in columns 3–48.
- Using a 1536 pintool, 20nl of 2mM compound are added to each well in columns 5–48.
- 20nl of DMSO are added to the wells in columns 1–4.
- 1.5ul of assay buffer containing 450µM PEA is added to all wells.
- The plate is centrifuged and incubated at room temperature for 1 hour.
- 3ul of Biomol Green is added to each well.
- The plate is centrifuged and incubated for 30 min at room temperature.
- The plate is read on a Viewlux plate reader in absorbance mode @630nm.
The average Z’ for the screen was 0.56, the signal-to-background was 11.5, signal-to-noise was 21.8 and signal-to-window was 6.6.
Rationale for confirmatory, counter and selectivity assays
Compounds that looked promising based on their selectivity profile were then tested in dose-response secondary assays using 10-point serial dilution in duplicate. The assay was based on detection of phosphate release from phosphoethanolamine using the Biomol reagent. We also developed another secondary assay for PHOSPHO1 which was based on the detection of choline released in the dephosphorylation of phosphocholine, an endogenous substrate for PHOSPHO1. Phosphate was also detected with the Biomol reagent (see Table 2).
Table 2
Assays used in PHOSPHO1 studies.
Knowledge of specificity is important for the delineation of PHOSPHO1 biological functions, specifically the selectivity of inhibitors for PHOSPHO1 versus TNAP present in the same tissue and participating in similar biological processes.
iii. Summary of Results
The uHTS assay was developed and implemented in 1536-well plates. After successful implementation the assay underwent uHTS with a library of ~55K compounds. Approximately 5000 compounds showed >50% activity in the assay at that point. This gave a hit rate of ~ 3%, which was unexpectedly high and difficult to follow up. Interestingly, more than 50% of the hits on two representative plates that were cherry-picked using our acoustic Echo dispenser confirmed in both single-concentration and dose-response modes.
During the preparation for this probe project we realized that PHOSPHO1, an orphan phosphatase with little information available, belongs to a family of halo-acid dehydrogenases. Thus, it was expected to share some common features with other members of the same family, including phosphomannomutase-2 (PMM2). We had already developed an assay for PMM2 and utilized it as a counter screen for phosphomannoseisomerase (PMI), an MLSCN probe project. We also noticed that in the PHOSPHO1R03 grant application three compounds had previously been identified as potential hits. We had already demonstrated that one of them (ebselen) inhibits both PMI and PMM2. Since we had previously generated analogues by purchase and synthesis we had a large number of dry powder compounds based on this scaffold available for testing. We therefore decided to test these compounds against PHOSPHO1 and found that several of them were very potent and selective for PHOSPHO1, not PMM or PMI active, but also satisfyingly not active against TNAP, the original anti-target of most concern for PHOPHOSPHI activity.
c. Probe Optimization
i. SAR & chemistry strategy that led to the probe
The goals for optimization of the PHOSPHO1 hit compounds from the primary HTS screen were two-fold: (1) find analogues with increased potency in the PHOSPHO1 enzyme inhibition assay and (2) optimize the potent PHOSPHO1 inhibitors for selectivity against TNAP. Additionally, as it was realized that PHOSPHO1 was a relative of the haloacid dehydrogenases, PMM2 and PMI, therefore, we also included 2 specifity assays for the class of ebselen-like inhibitors we had previously tested for: PMM and PMI enzyme inhibition, and indeed we used these to actually select and define the final desired compound, since TNAP activity was minimal for these series (see below and Table 3). HTS revealed multiple potent hits in the benzoisothiazolone compound class, with a majority of the inhibitors containing a sulfonamide group as in CID-4089709 (MLS-0315803), CID-2325813 (MLS-0315794), CID-5040456 (MLS-0263839), and CID-2327953 (MLS-0022297) (Table 3). Select representatives of these potent hits were re-synthesized in house and activity was confirmed with synthesized analogues. All of the data for these analogs and the probe were obtained from powder samples from the same batch for each replicate experiments. While the sulfonamide compounds were indeed potent PHOSPHO1 inhibitors, counterscreening against TNAP, PMM and PMI enzymes showed most to be non-selective. For example, CID-4089709 (MLS-0315803) showed an IC50 of 0.81 µM against PHOSPHO1. However, this compound inhibited PMM with an IC50 of 5.17 µM and was even more potent against PMI with an IC50 of 0.71 µM. Thus, the SAR strategy for medicinal chemistry included variations of the sulfonamide moiety on the aryl ring as well as replacing the sulfonamide with suitable, drug-like isosteres, specifically amide and ester analogues. Regarding the SAR around the sulfonamide analogues, it was observed that large sulfonamide substituents, as in CID-4089709 (MLS-0315803), whilst proving to be potent at inhibiting PHOSPHO1, also showed activity against PMM and PMI. Thus, the focus shifted to smaller substituents. Selectivity began to emerge as these smaller, less polar substituents were incorporated, as in CID-5040456 (MLS-0263839), CID-2327953 (MLS-0022297), and CID-2325813 (MLS-0315794), all of which had PHOSPHO1 selectivities from 4- to 100-fold over both PMM and PMI. However, additional selectivity for PHOSPHO1 was sought. Gratifyingly, CID-2381612 (MLS-0315805), which had an IC50 of 0.56 µM against PHOSPHO1, showed no inhibition against TNAP and PMM up to 100 µM, and a PMI IC50 of 61.9 µM. While this compound demonstrated desirable potency and selectivity, we envisaged that potency could be further increased since we had previously observed non-selective compounds that were 2- to 3-fold more potent against PHOSPHO1. Encouraging potency and selectivity results were seen when the sulfonamide group was replaced with either an amide or ester isostere. As was previously observed with the sulfonamide series, larger substituents, as in CID-16749997 (MLS-0390837), were potent (PHOSPHO1 IC50 = 2.33 µM), yet lacked the required selectivity against PMM and PMI. Subsequent synthesis of esters and amides containing smaller substituents facilitated a means to achieve the required selectivity while maintaining potency, as shown in CID-2381612 (MLS-0315805). Thus, a final round of synthesis of amides afforded the probe compound. CID-16749996 MLS-0390838) proved to be the most potent and selective PHOSPHO1 compound synthesized, having an IC50 of 0.14 µM and negligible inhibition of PMM (IC50 = 76.4 µM) or TNAP and PMI (IC50> 100 µM).
Table 3
Representative PHOSPHO1 inhibitors: SAR and selectivity.
3. Probe
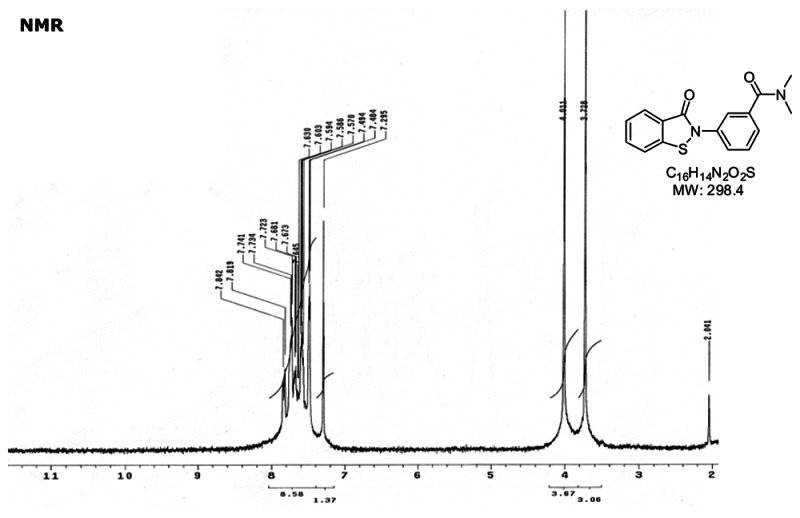
a. Chemical name
N,N-dimethyl-3-(3-oxo-1,2-benzothiazol-2-yl)benzamide [ML086]
b. Probe chemical structure


d. PubChem CID (corresponding to the SID)
CID-16749996
e. Availability from a vendor
This probe is not commercially available from vendors.
f. MLS#'s of probe molecule and five related samples that were submitted to the SMR collection
| Probe/Analog | MLS-# (BCCG#) | CID | SID | Source (vendor or BCCG syn) | Amt (mg) | Date ordered/submitted |
|---|---|---|---|---|---|---|
| Probe | 0390838 | 16749996 | 57287582 | BCCG syn | 20 | 4/15/09 |
| Analog 1 | 0390837 | 16749997 | 57287581 | BCCG syn | 20 | 4/15/09 |
| Analog 2 | 0315861 | 25067463 | 56405542 | BCCG syn | 20 | 4/15/09 |
| Analog 3 | 0263839 | 5040456 | 56373499 | Enamine | 20 | 4/15/09 |
| Analog 4 | 0022297 | 2327953 | 56373866 | Enamine | 20 | Backorder |
| Analog 5 | 0315803 | 4089709 | 56373872 | Enamine | 20 | Backorder |
g. Mode of action for biological activity of probe
CID-16749996 is a biochemical inhibitor of PHOSPHO1. The biological mode of action of this probe has not yet been elucidated.
h. Detailed synthetic pathway for making probe
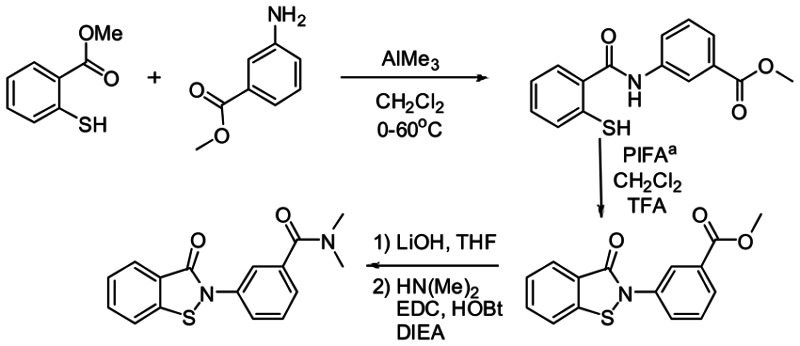
Synthesis of CID-16749996.N,N-dimethyl-3-(3-oxobenzo[d]isothiazol-2(3H)-yl)benzamide.
i. Summary of probe properties (solubility, absorbance/fluorescence, reactivity, toxicity, etc.)
No obvious reactivities or toxicities.
j. Properties Computed from Structure
| Property | Value |
|---|---|
| Molecular Weight | 298.35956 [g/mol] |
| Molecular Formula | C16H14N2O2S |
| XLogP3-AA | 2.5 |
| H-Bond Donor | 0 |
| H-Bond Acceptor | 2 |
| Rotatable Bond Count | 2 |
| Exact Mass | 298.077598 |
| MonoIsotopic Mass | 298.077598 |
| Topological Polar Surface Area | 65.9 |
| Heavy Atom Count | 21 |
| Formal Charge | 0 |
| Complexity | 427 |
| Isotope Atom Count | 0 |
| Defined Atom StereoCenter Count | 0 |
| Undefined Atom StereoCenter Count | 0 |
| Defined Bond StereoCenter Count | 0 |
| Undefined Bond StereoCenter Count | 0 |
| Covalently-Bonded Unit Count | 1 |
4. Appendices
a. Comparative data on (1) probe, (2) similar compound structures (establishing SAR) and (3) prior probes
The three compounds (Fig. 1A) on the left were obtained during pilot screening of two well-known chemical libraries, LOPAC and Spectrum. Their calculated IC50 values against PHOSPHO1-mediated hydrolysis of PEA were 1.97 ± 0.01 μM (SCH 202676), 4.71 ± 0.1 μM (Lansoprazole) and 2.81 ± 0.04 μM (Ebselen) (Fig. 1B).
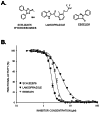
None of these meet the current minimal definition of less than 1 µM potency for a biochemical assay. The nominated probe meets the potency requirement and also meets the original need for TNAP selectivity. It also exceeds the specification with the added selectivity against two related in-family enzyme members. However, the biological relevance of these homologous enzyme to calcification is not known. The probe provided has however does have this added selectivity, which presumably minimize potential off-target effects.
b. Comparative data showing probe specificity for target
See Table 3 above for selectivity.
5. Bibliography
- 1.
- Millán JL. Mammalian alkaline phosphatases From biology to applications in medicine and biotechnology. Wiley-VCH Verlag GmbH & Co; Weinheim, Germany: 2006. pp. 1–322.
- 2.
- Whyte MP. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev. 1994;15:439–461. [PubMed: 7988481]
- 3.
- Henthorn PS, Raducha M, Fedde KN, Lafferty MA, Whyte MP. Different missense mutations at the tissue-nonspecific alkaline phosphatase gene locus in autosomal recessively inherited forms of mild and severe hypophosphatasia. Proc Natl Acad Sci USA. 1992;89:9924–8. [PMC free article: PMC50246] [PubMed: 1409720]
- 4.
- Fukushi M, Amizuka N, Hoshi K, Ozawa H, Kumagai H, Omura S, Misumi Y, Ikehara Y, Oda K. Intracellular retention and degradation of tissue-nonspecific alkaline phosphatase with a Gly317-->Asp substitution associated with lethal hypophosphatasia. Biochem. Biophys. Res. Comm. 1998;246:613–618. [PubMed: 9618260]
- 5.
- Shibata H, Fukushi M, Igarashi A, Misumi Y, Ikehara Y, Ohashi Y, Oda K. Defective intracellular transport of tissue-nonspecific alkaline phosphatase with an Ala162-->Thr mutation associated with lethal hypophosphatasia. J Biochem. 1988;123:968–977. [PubMed: 9562633]
- 6.
- Zurutuza L, Muller F, Gibrat JF, Taillandier A, Simon-Bouy B, Serre JL, Mornet E. Correlations of genotype and phenotype in hypophosphatasia. Hum Mol Genet. 1999;8:1039–1046. [PubMed: 10332035]
- 7.
- Whyte MP. Hypophosphatasia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 7th ed. McGraw-Hill; New York: 1995. pp. 4095–4112.
- 8.
- Narisawa S, Fröhlander N, Millán JL. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev Dyn. 1997;208:432–446. [PubMed: 9056646]
- 9.
- Fedde KN, Blair L, Silverstein J, Coburn SP, Ryan LM, Weinstein RS, Waymire K, Narisawa S, Millan JL, MacGregor GR, Whyte MP. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res. 1999;14:2015–26. [PMC free article: PMC3049802] [PubMed: 10620060]
- 10.
- Ali SY, Sajdera SW, Anderson HC. Isolation and characterization of calcifying matrix vesicles from epiphyseal cartilage. Proc Natl Acad Sci USA. 1970;67:1513–1520. [PMC free article: PMC283384] [PubMed: 5274475]
- 11.
- Bernard GW. Ultrastructural localization of alkaline phosphatase in initial membranous osteogenesis. Clin Orthop. 1978;135:218–225. [PubMed: 709934]
- 12.
- Morris DC, Masuhara K, Takaoka K, Ono K, Anderson HC. Immunolocalization of alkaline phosphatase in osteoblasts and matrix vesicles of human fetal bone. Bone Miner. 1992;19:287–98. [PubMed: 1472898]
- 13.
- Robison R. The possible significance of hexosephosphoric esters in ossification. Biochem J. 1923;17:286–293. [PMC free article: PMC1259346] [PubMed: 16743183]
- 14.
- Majeska RJ, Wuthier RE. Studies on matrix vesicles isolated from chick epiphysealcartilage. Association of pyrophosphatase and ATPase activities with alkaline phosphatase. Biochim Biophys Acta. 1975;391:51–60. [PubMed: 237558]
- 15.
- Fallon MD, Whyte MP, Teitelbaum SL. Stereospecific inhibition of alkaline phosphatase by Ltetramisole prevents in vitro cartilage calcification. Lab Invest. 1980;43:489–494. [PubMed: 6449630]
- 16.
- Meyer JL. Studies on matrix vesicles isolated from chick epiphyseal cartilage. Association of pyrophosphatase and ATPase activities with alkaline phosphatase. Arch Biochem Biophys. 1984;231:1–8. [PubMed: 237558]
- 17.
- Moss DW, Eaton RH, Smith JK, Whitby LG. Association of inorganic pyrophosphatase activity with human alkaline phosphatase preparations. Biochem J. 1967;102:53–57. [PMC free article: PMC1270209] [PubMed: 6030299]
- 18.
- Rezende A, Pizauro J, Ciancaglini P, Leone F. Phosphodiesterase activity is a novel property of alkaline phosphatase from osseous plate. Biochem J. 1994;301:517–522. [PMC free article: PMC1137111] [PubMed: 8042997]
- 19.
- Anderson HC, Garimella R, Tague SE. The role of matrix vesicles in growth plate development and biomineralization. Frontiers in Bioscience. 2005;10:822–837. [PubMed: 15569622]
- 20.
- Anderson HC, Sipe JE, Hessle L, Dhamayamraju R, Atti E, Camacho NP, Millán JL. Impaired calcification around matrix vesicles of growth plate and bone in alkaline phosphatase-deficient mice. Am J Pathol. 2004;164:841–847. [PMC free article: PMC1613274] [PubMed: 14982838]
- 21.
- Terkeltaub RA. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol. 2001;281:C1–11. [PubMed: 11401820]
- 22.
- Huang R, Rosenbach M, Vaughn R, Provvedini D, Rebbe N, Hickman S, Goding J, Terkeltaub R. Expression of the murine plasma cell nucleotide pyrophosphohydrolase PC-1 is shared by human liver, bone, and cartilage cells. Regulation of PC-1 expression in osteosarcoma cells by transforming growth factor-beta. J Clin Invest. 1994;94:560–567. [PMC free article: PMC296131] [PubMed: 8040311]
- 23.
- Bollen M, Gijsbers R, Ceulemans H, Stalmans W, Stefan C. Nucleotide pyrophophatasesphosphodiesterases on the move. Crit Rev Biochem Mol Biol. 2000;35:393–432. [PubMed: 11202013]
- 24.
- Meyer JL. Studies on matrix vesicles isolated from chick epiphyseal cartilage. Association of pyrophosphatase and ATPase activities with alkaline phosphatase. Arch Biochem Biophys. 1984;231:1–8. [PubMed: 237558]
- 25.
- Hashimoto S, Ochs RL, Rosen F, Quach J, McCabe G, Solan J, Seegmiller JE, Terkeltaub R, Lotz M. Chondrocyte-derived apoptotic bodies and calcification of articular cartilage. Proc Natl Acad Sci USA. 1998;95:3094–3099. [PMC free article: PMC19700] [PubMed: 9501221]
- 26.
- Johnson K, Moffa A, Chen Y, Pritzker K, Goding J, Terkeltaub R. Matrix vesicle plasma membrane glycoprotein-1 regulates mineralization by murine osteoblastic MC3T3 cells. J Bone Miner Res. 1999;14:883–892. [PubMed: 10352096]
- 27.
- Terkeltaub R, Rosenbach M, Fong F, Goding J. Causal link between nucleotide pyrophosphohydrolase overactivity and increased intracellular inorganic pyrophosphate generation demonstrated by transfection of cultured fibroblasts and osteoblasts with plasma cell membrane glycoprotein-1. Relevance to clacium pyrophosphate dihydrate deposition disease. Arthritis Rheum. 1994;37:934–941. [PubMed: 8003067]
- 28.
- Johnson K, Moffa A, Chen Y, Pritzker K, Goding J, Terkeltaub R. Matrix vesicle plasma membrane glycoprotein-1 regulates mineralization by murine osteoblastic MC3T3 cells. J Bone Miner Res. 1999;14:883–892. [PubMed: 10352096]
- 29.
- Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y, Ikegawa S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat Genet. 1998;19:271–3. [PubMed: 9662402]
- 30.
- Sali A, Favaloro JM, Terkeltaub R, Goding JW. Germline deletion of the nucleoside triphosphatepyrophosphohydrolase (NTPPPH) plasma cell membrane glycoprotein (PC-1) produces abnormal calcification of periarticular tissues. Maastricht: Shaker Publishing BV; 1999.
- 31.
- Johnson K, Hashimoto S, Lotz M, Pritzker K, Goding J, Terkeltaub R. Up-regulated expression of the phosphodiesterase nucleotide pyrophosphatase family member PC-1 is a marker and pathogenic factor for knee meniscal cartilage matrix calcification. Arthr Rheum. 2001;44:1071–1081. [PubMed: 11352238]
- 32.
- Masuda I, Iyama K, Halligan BD, Barbieri JT, Haas AL, McCarty DJ, Ryan LM. Variations in site and levels of expression of chondrocyte nucleotide pyrophosphohydrolase with aging. J Bone Miner Res. 2001;16:868–875. [PubMed: 11341331]
- 33.
- Jones AC, Chucjk AJ, Arie EA, Green DJ, Doherty M. Diseases associated with calcium pyrophosphate deposition disease. Semin Arthritis Rheum. 1992;22:188–202. [PubMed: 1295092]
- 34.
- Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289:265–70. [PubMed: 10894769]
- 35.
- Nürnberg P, Theile H, Chandler D, Höhne W, Cunningham ML, Ritter H. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat Genet. 2001;28:37–41. [PubMed: 11326272]
- 36.
- Wang W, Xu J, Du B, Kirsch T. Role of the Progressive Ankylosis Gene (ank) in Cartilage Mineralization. Mol Cell Biol. 2005;25:312–323. [PMC free article: PMC538760] [PubMed: 15601852]
- 37.
- Sweet HO, Green MC. Progressive ankylosis, a new skeletal mutation in the mouse. J Hered. 1981;72:87–93. [PubMed: 7276519]
- 38.
- Hakim FT, Cranley R, Brown KS, Eanes ED, Harne L, Oppenheim JJ. Hereditary joint disorder in progressive ankylosis (ank/ank) mice. I. Association of calcium hydroxyapatite deposition with inflammatory arthropathy. Arthritis Rheum. 1984;27:1411–1420. [PubMed: 6095872]
- 39.
- Sampson HW, Davis JS. Histopathology of the intervertebral disc of progressive ankylosis mice. Spine. 1988;13:650–654. [PubMed: 3175756]
- 40.
- Mahowald ML, Krug H, Halverson J. Progressive ankylosis (ank/ank) in mice: an animal model of spondyloarthropathy. II. Light and electron microscopic findings. J Rheumatol. 1989;16:60–66. [PubMed: 2541245]
- 41.
- Balsan S, Garabedian M, Larchet M, Gorski AM, Cournot G, Tau C, Bourdeau A, Silve C, Ricour C. Long-term nocturnal calcium infusions can cure rickets and promote normal mineralization inheriditary resistance to 1, 25-Dihydroxyvitamin D. J Clin Invest. 1986;77:1661–1667. [PMC free article: PMC424571] [PubMed: 3009551]
- 42.
- Hessle L, Johnsson KA, Anderson HC, Narisawa S, Sali A, Goding JW, Terkeltaub R, Millán JL. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci USA. 2002;99:9445–9449. [PMC free article: PMC123160] [PubMed: 12082181]
- 43.
- Harmey D, Hessle L, Narisawa S, Johnson K, Terkeltaub R, Millán JL. Concerted regulation of inorganic pyrophosphate and osteopontin by Akp2, Enpp1 and Ank. An integrated model of the pathogenesis of mineralization disorders. Am J Pathol. 2004;164:1199–1209. [PMC free article: PMC1615351] [PubMed: 15039209]
- 44.
- Stewart AJ, Schmid R, Blindauer CA, Paisley SJ, Farquharson C. Comparative modelling of human PHOSPHO1 reveals a new group of phosphatases within the HAD superfamily. Protein Engineering. 2003;16:889–895. [PubMed: 14983068]
- 45.
- Houston B, Stewart AJ, Farquharson C. PHOSPHO1 - A novel phosphatase specifically expressed at sites of mineralization in bone and cartilage. Bone. 2004;34:629–637. [PubMed: 15050893]
- 46.
- Roberts SJ, Stewart AJ, Sadler PJ, Farquharson C. Human PHOSPHO1 displays high specific phosphoethanolamine and phosphocholine phosphatase activity. Biochem J. 2004;382:59–65. [PMC free article: PMC1133915] [PubMed: 15175005]
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: a unified model of the mechanisms of initiation of skeletal calcification.[J Bone Miner Res. 2011]Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: a unified model of the mechanisms of initiation of skeletal calcification.Yadav MC, Simão AM, Narisawa S, Huesa C, McKee MD, Farquharson C, Millán JL. J Bone Miner Res. 2011 Feb; 26(2):286-97. Epub 2010 Aug 3.
- Review The role of phosphatases in the initiation of skeletal mineralization.[Calcif Tissue Int. 2013]Review The role of phosphatases in the initiation of skeletal mineralization.Millán JL. Calcif Tissue Int. 2013 Oct; 93(4):299-306. Epub 2012 Nov 27.
- Perspective on Dentoalveolar Manifestations Resulting From PHOSPHO1 Loss-of-Function: A Form of Pseudohypophosphatasia?[Front Dent Med. 2022]Perspective on Dentoalveolar Manifestations Resulting From PHOSPHO1 Loss-of-Function: A Form of Pseudohypophosphatasia?Mohamed FF, Chavez MB, de Oliveira FA, Narisawa S, Farquharson C, Millán JL, Foster BL. Front Dent Med. 2022 Feb; 3. Epub 2022 Feb 3.
- Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralization.[J Bone Miner Res. 2007]Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralization.Roberts S, Narisawa S, Harmey D, Millán JL, Farquharson C. J Bone Miner Res. 2007 Apr; 22(4):617-27.
- Review Role of bone-type tissue-nonspecific alkaline phosphatase and PHOSPO1 in vascular calcification.[Curr Pharm Des. 2014]Review Role of bone-type tissue-nonspecific alkaline phosphatase and PHOSPO1 in vascular calcification.Bobryshev YV, Orekhov AN, Sobenin I, Chistiakov DA. Curr Pharm Des. 2014; 20(37):5821-8.
- The Role of PHOSPHO1 in the Initiation of Skeletal Calcification - Probe Reports...The Role of PHOSPHO1 in the Initiation of Skeletal Calcification - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...