

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

A nitrile-containing propionic acid moiety as an electrophile for covalent attack by the active site cysteine residue of caspase 1 was investigated. Several cyanopropanate containing small molecules were synthesized, including one based upon the optimized peptidic scaffold of the prodrug VX-765. A number of these compounds were potent inhibitors of caspase 1 (IC50s ≤ 1 nM). Examination of these small molecules versus a caspase panel demonstrated an impressive degree of selectivity for caspase 1 inhibition. The small molecular probe ML132 (CID-4462093; NCGC-00183434) is the most potent caspase 1 inhibitor reported to date. It also possesses a unique selectivity pattern relative to other reported caspase inhibitors. A number of these compounds were assessed for their hydrolytic stability and selected absorption, distribution, metabolism and elimination (ADME) properties.
Assigned Assay Grant #: X01 MH078950-01
Screening Center Name & PI: NIH Chemical Genomics Center & Dr. Christopher P. Austin
Chemistry Center Name & PI: NIH Chemical Genomics Center & Dr. Christopher P. Austin
Assay Submitter & Institution: Dr. Jim Wells, University of California at San Francisco
PubChem Summary Bioassay Identifier (AID): 2389
Probe Structure & Characteristics

Recommendations for scientific use of the probe
Caspase 1, also known as interleukin-converting enzyme or ICE, is responsible for the proteolytic activation of interleukin (IL)-1β and IL-18. [1] IL-1β and IL-18 are cytokines that play a major role in the immune response and within numerous autoimmune and inflammatory diseases. Caspase 1 is constitutively and inducibly expressed in immune response elements such as T cells, macrophages and neutrophils. Inhibitors of caspase 1 are sought for intervention strategies within ischemic disorders, Huntington’s disease, amyotrophic lateral sclerosis (ALS), rheumatoid arthritis, osteoarthritis, inflammatory bowel disease and sepsis.
1. Introduction
Caspases are cysteine proteases with a strict specificity for cleaving peptide sequences C-terminal to aspartic acids residues. Currently, 12 caspase isozymes have been identified in humans, with numerous reported activities. Caspases are often subcategorized as either pro-apoptotic or pro-inflammatory enzymes. A prominent member of the pro-inflammatory class is caspase 1, also known as interleukin-converting enzyme or ICE, which is responsible for the proteolytic activation of interleukin (IL)-1β and IL-18 [1]. IL-1β and IL-18 are cytokines that play a major role in the immune response and within numerous autoimmune and inflammatory diseases [2]. Caspase 1 is constitutively and inducibly expressed in immune response elements such as T cells, macrophages and neutrophils. Procaspase 1 is known to associate with several multi-protein complexes capable of responding to numerous external stimuli, suggesting that caspase 1 is a major regulator of the inflammation response. Targeting proteases, and specifically caspases, via small molecule therapeutics is an active area of research. Small molecule inhibitors of selected proteases have entered the clinic, and many have received approval. Inhibitors of caspase 1 are sought for intervention strategies within ischemic disorders, Huntington’s disease, amyotrophic lateral sclerosis (ALS), rheumatoid arthritis, osteoarthritis, inflammatory bowel disease and sepsis.
The overall goal of this project is to screen for and/or design inhibitors of caspase 1 that are potent and selective over the 11 additional caspase isozymes. To date, at least three caspase 1 inhibitors have entered clinical evaluation, including Pralnacasan (VX-740), IDN-6556 and VX-765. All three agents are active site inhibitors that act through reversible (Pralnacasan and VX-765) or irreversible (IDN-6556) covalent modification of the catalytic cysteine residue. These agents have impressive potency versus caspase 1 (for instance, VX-740 has a published Ki of 1 nM) and good reported selectivity [3]. Any probe from this project must be an improvement on current art (i.e. potency better than VX-765 and equal selectivity). These agents, and the probes found herein, are covalent modifiers of their target. Often, covalent modifiers are avoided due to promiscuity issues and potential for toxicities within in vivo studies. However, a large number of known drugs do indeed inhibit their target via covalent modification at the orthosteric site, and as tool compounds, covalent modifiers can be very useful if their selectivity can be shown versus related targets.
2. Materials and Methods
General Methods for Chemistry
All air or moisture sensitive reactions were performed under positive pressure of nitrogen with oven-dried glassware. Anhydrous solvents such as dichloromethane, N, N-dimethylforamide (DMF), acetonitrile, methanol and triethylamine were obtained by purchasing from Sigma-Aldrich. Preparative purification was performed on a Waters semi-preparative HPLC. The column used was a Phenomenex Luna C18 (5 micron, 30 × 75 mm) at a flow rate of 45 mL/min. The mobile phase consisted of acetonitrile and water (each containing 0.1% trifluoroacetic acid). A gradient of 10% to 50% acetonitrile over 8 minutes was used during the purification. Fraction collection was triggered by UV detection (220 nM). Analytical analysis was performed on an Agilent LC/MS (Agilent Technologies, Santa Clara, CA).
Method 1: A 7 minute gradient of 4% to 100% Acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with an 8 minute run time at a flow rate of 1 ml/min. A Phenomenex Luna C18 column (3 micron, 3 × 75 mm) was used at a temperature of 50°C.
Method 2: A 3 minute gradient of 4% to 100% Acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 4.5 minute run time at a flow rate of 1 mL/min. A Phenomenex Gemini Phenyl column (3 micron, 3 × 100 mm) was used at a temperature of 50 °C.
Purity determination was performed using an Agilent Diode Array Detector on both Method 1 and Method 2. Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. 1H NMR spectra were recorded on Varian 400 MHz spectrometers. Chemical Shifts are reported in ppm with tetramethylsilane (TMS) as internal standard (0 ppm) for CDCl3 solutions or undeuterated solvent (DMSO-h6 at 2.49 ppm) for DMSO-d6 solutions. All of the analogs for assay have purity greater than 95% based on both analytical methods. High resolution mass spectrometry was recorded on an Agilent 6210 Time-of-Flight LC/MS system. Confirmation of molecular formula was accomplished using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02).
2.1. Assays
The biochemical assay was configured using purified caspase 1 at a high enzyme concentration to promote formation of the homodimers, and thus facilitate the identification of allosteric inhibitors. The overall goal was to identify reversible inhibitors with better drug-like properties than the currently available set of aspartyl-containing peptidomimetics that covalently bind the active site. Caspase 1 was assayed using the profluorescent substrate Ac-WEHD-AFC. After initiation of the assay with substrate, the plates were rapidly read using an automated robotic system (Kalypsys, Inc.) to maintain consistent timing. A kinetic mode of detection was used where the initial rate was collected (estimated final product formation was ~10%). Compounds were screened as a concentration-titration series that ranged from 57μM to 3.7nM. Below is the protocol used for caspase 1.
Caspase 1 was prepared in buffer (50mM HEPES pH 7.5, 50mM KCl, 200mM NaCl, 10mM DTT, 0.1% CHAPS) at a concentration of 66.6nM, and 3μl was dispensed to all wells using black solid Kalypsys 1536-well plates. 20nl of DMSO containing compounds was added using a Kalypsys pin-tool to columns 5–48. Then, 20nl of DMSO solution from a control plate was added to columns 1–4. Controls were: Column 1, 16 point titration with each concentration in duplicate (1:1 dilutions in DMSO; final starting concentration was 57μM) of the caspase 1 inhibitor Ac-WEHD-CHO (Alexis Biochemicals); Column 2, a 16 point titration with each concentration in duplicate of the free AFC fluorophore prepared in DMSO (Alexis Biochemicals), final starting concentration was 40μM; Column 3 neutral (DMSO only) control; Column 4: DMSO alone, to serve as a negative control (no substrate was added). Then 1μl of 20μM of the substrate Ac-WEHD-AFC (Alexis Biochemicals) prepared in the same buffer was dispensed to all wells except columns 2 and 4, and the plates were immediately transferred (<1 min) to the Viewlux. The plates were then exposed using 405 nm excitation/520 nm emission filters for 4 sec and read at 20 sec intervals for 3 min. Final enzyme concentration was 50 nM and the final substrate concentration was 5μM.
Concentration-response curves were fitted to the data calculated from slope of the linear regression of fluorescent intensity versus time (the rate). The concentration-effect curves were then classified based on curve quality (r2), response magnitude and degree of measured activity. Active compounds showed concentration-dependent decreases in the measured rate. Inconclusive compounds had appreciable concentration-dependent effects on both the measured rate and the interpolated basal fluorescence intensity at the start of the reaction, but where the basal fluorescent intensity was marginal (e.g. < 10-fold). Inconclusive compounds also encompass highly fluorescent compounds (> 100-fold increases in fluorescent intensities values relative to controls) where artificial concentration-dependent decreases in the rate occurred at high compound concentrations due to fluorescent interferences.
The assay was well behaved; the signal-to-background ratio was on average 13 and the average Z′ screening factor associated with each plate was 0.8, indicating a robust performance of the screen. A total of 20 actives were found and submitted. None of these agents had the requirements to satisfy the probe definition. Additional assays were performed by commercial vendor (www.reactionbiology.com); these assays rely upon a peptide cleavage assay of a caged florophore (Z-LEHD)2-R110 tetrapeptide.
2.2. Probe Chemical Characterization

Scheme 1Conditions and reagents
(a) EDC, DMAP, CH2Cl2, 6 h(59%); (b) TFAA, DIPEA, CH2Cl2, 0°C, 30 min. (90%); (c) thionyl chloride (excess), EtOH, 0°C (95%); (d) TFAA, DIPEA, CH2Cl2, 0°C, 30 min. (86%); (e) NMM, DME, then NH4OH (73%); (f) TMSN3, Bu2SnO (0.6 equiv.), toluene, μW, 100°C, 1 h (77%); (g) TFAA, DIPEA, CH2Cl2, 0°C, 30 min. (80%).
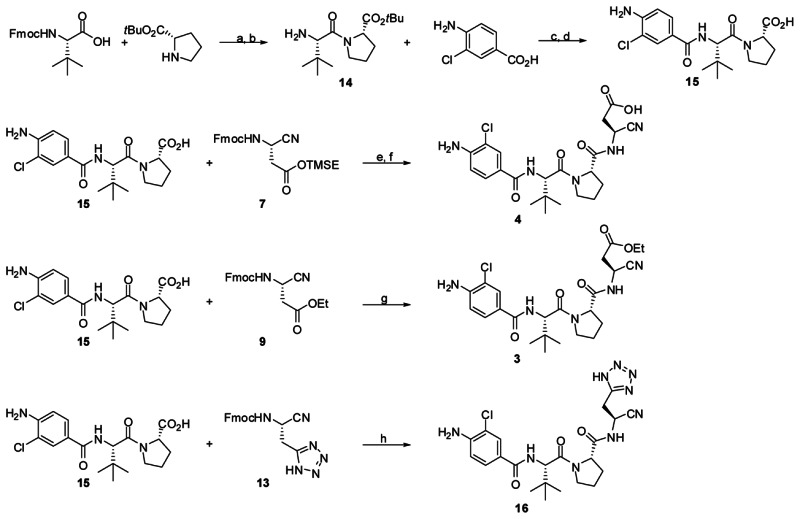
Scheme 2Conditions and reagents
(a) EDC, HOBt, DMF, rt, 8 h; (b) DBU, CH2Cl2, rt (71% over 2 steps); (c) HATU, DIPEA, DMF, rt, 2 h; (d)TFA,CH2Cl2 (1:1), rt, 4 h (85% over 2 steps); (e) DBU, DMF, 5 min. then 15, HATU, DIPEA, DMF, 0 °C, 2h; (f) TBAF, THF, 0 °C (72% over 2 steps); (g) DBU, DMF, 5 min. then 15, HATU, DIPEA, DMF, 0 °C, 2h (91%); (h) DBU, DMF, 5 min. then 15, HATU, DIPEA, DMF, 0 °C, 2h (82%).
NCGC-00183434/CID-44620939/ML132: 1H NMR (400 MHz, DMSO-d6) δ ppm 8.68 (d, J=7.4 Hz, 1 H), 7.79 (d, J=1.9 Hz, 1 H), 7.64 (d, J=9.0 Hz, 1 H), 7.56 (dd, J=8.6, 1.9 Hz, 1 H), 6.73 (d, J=8.6 Hz, 1 H), 5.95 (b, 2H), 4.83 (q, J=7.4 Hz, 1H), 4.64 (d, J=9.0 Hz, 1 H), 4.20 – 4.30 (dd, J= 8.2, 5.6 Hz 1 H), 3.69 – 3.80 (m, 1 H), 3.60 (m, 1 H), 2.79 (dd, J=6.7, 3.1 Hz, 2 H), 1.99 – 2.11 (m, 1 H), 1.63 – 1.96 (m, 4 H), 0.99 (s, 9 H) 13C NMR (100 MHz, DMSO-d6): 172.2, 170.6, 169.9, 165.8, 148.0, 129.4, 128.2, 122.1, 119.1, 116.4, 114.4, 59.7, 57.8, 48.2, 37.4, 36.6, 35.3, 29.5, 27.0, 25.1. LC/MS: Method 1, retention time: 4.595 min; Method 2, retention time: 3.591 min; HRMS: m/z (M+H+) = 477.1784 (Calculated for C22H28N5O5Cl = 477.1779). [α]22D = −65 (c 1.0, MeOH). Solubility (PBS, pH 7.4, 23°C) ≥ 100μM. Stability profile over 100 hrs (water, pH 7, pH 2 and pH 8, 23°C) is shown in Figure 1.

2.3. Probe Preparation
Appropriately substituted ethyl-3-cyanopropanoate derivatives are not commercially available, and therefore required synthetic elaboration. As we desired to explore both an active and prodrug form of our conceived molecule, we examined alternative protecting group strategies for the acid side chain (Scheme 1). Commercially available Fmoc protected D-isoasparagine (5) offered a convenient entry point to both required building blocks. Treatment of 5 with 2-(trimethylsilyl)ethanol, EDC and DMAP in methylene chloride provided the TMSE protected 6 in good yield. Conversion of 6 to nitrile 7 was accomplished by treatment with trifluoroacetic anhydride and Hunig’s base. A similar sequence was used to produce the ethyl ester 9. In addition to the ester prodrug and the active cyanopropionic acid, it was of interest to explore carboxylic acid mimetics. As such, we undertook the synthesis of a tetrazole version of the key ethyl-3-cyanopropanoate moiety. Here, we utilized the previously reported Fmoc protected (S)-2-amino-3-cyanopropanoic acid (10). Conversion to amide 11 was required prior to formation of the tetrazole 12. The amide was formed via the mixed anhydride, followed by treatment with ammonium hydroxide. Tetrazole formation was accomplished via microwave irradiation of the nitrile 11 and TMS-azide in the presence of dibutylstannanone. Dehydration to nitrile 13 was accomplished in a manner analogous to 7 and 9. With appropriately substituted/protected cyanopropanoate building blocks, we next turned our attention to the trimer core of VX-765 (Scheme 2). Both Fmoc protected L-tert-leucine and tert-butyl-L-prolinate are commercially available and were easily coupled via treatment with EDC and HOBt. Fmoc removal was effected by treatment with DBU resulting in the protected dimer 14. Coupling of 14 with 4-amino-3-chlorobenzoic acid was accomplished using HATU and Hunig’s base in DMF. TFA mediated removal of the tert-butyl group yielded the carboxylic acid 15. A single pot deprotection-coupling sequence was used to generate the desired final products. Treatment of 7, 9 and 13 with DBU in DMF effected deprotection to the free amines which were added sequentially to 15, Hunig’s base and finally HATU to yield the coupled products. The generation of 4 (NCGC-00183434/CID-44620939/ML132) further required TBAF mediated removal of the TMSE group.
3. Results
Please see subsection for a detailed description of the results.
3.1. Summary of Screening Results
Not applicable (these agents were rationally designed following our inability to discover new agents within the qHTS Assay for Allosteric/Competitive Inhibitors of Caspase-1; AID 900).
Identification of lead
The design of small molecule inhibitors of cysteine proteases relies heavily on covalent modification of the active site cysteine through reaction with the highly nucleophilic thiolate. Electrophilic ‘warhead’ moieties suitable for this modification include the aforementioned aldehyde, Michael acceptors (for instance, vinyl sulfones), α-halo ketones, epoxides and nitriles. In particular, nitrile-based cysteine protease inhibitors have found utility versus cathepsin K [4], TbCatB [5] and cruzain [6]. We endeavored to explore the promise of cyanopropanoates as caspase inhibitors (Figure 1B). To explore the potential of this functional moiety, we took advantage of the peptidic scaffold of VX-765. Further, we incorporated the ethyl-3- cyanopropanoate to mimic the prodrug qualities associated with VX-765 (Figure 1). VX-765 (1) is a prodrug that requires esterase cleavage of the 5-ethoxydihydrofuran-2(3H)-one moiety to yield the aldehyde functionality of the drug VRT-043198 (2b) (CID-11443029), which acts as a potent electrophile for attack by the active site cysteine thiol (Figure 1A). The remainder of the VX-765 (1) molecule establishes key binding contacts with caspase 1 that enhance the potency of the interaction and confer a modest degree of selectivity.
| Probe | NCGC-00183434 | MLS003178557 |
| Analog | NCGC-00241069 | MLS003178558 |
| Analog | NCGC-00241070 | MLS003178559 |
| Analog | NCGC-00241071 | MLS003178560 |
| Analog | NCGC-00185682 | MLS003178561 |
| Analog | NCGC-00241073 | MLS003178562 |

Figure 2
A. The structure of VX-765 (1) and schematic representation of esterase cleavage of the 5-ethoxydihydrofuran-2(3H)-one moiety to yield the active drug VRT-043198 (2b). B. The structure of NCGC-00185682 (3) and putative esterase cleavage of the ethyl-3-cyanopropanoate moiety to yield active agent NCGC-00183434/CID-44620939 (4).
3.2. Dose Response Curves for Probe
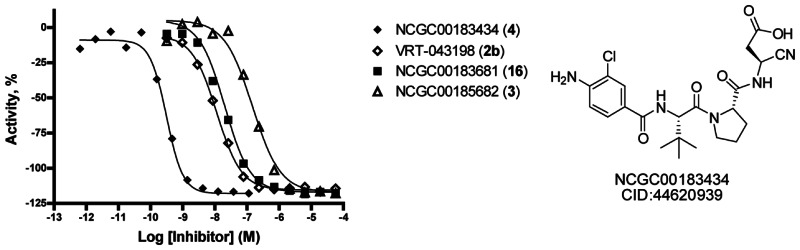
Figure 3Structure of NCGC-00183434/CID-44620939/ML132 and dose response curve for data acquired from the fluorescent enzyme assay using YVAD-CHO as a control and YVAD-AMC as the substrate
3.3. Scaffold/Moiety Chemical Liabilities
NCGC-00183434/CID-44620939/ML132 contains nitrile and acid moieties. The nitrile group is intended to be a covalent modifier of the target caspase 1. Often, covalent modifiers are avoided due to promiscuity issues and potential for toxicities within in vivo studies. However, a large number of known drugs do indeed inhibit their target via covalent modification at the orthosteric site, and as tool compounds, covalent modifiers can be very useful if their selectivity can be shown versus related targets. We have profiled these agents for selectivity versus within a commercial panel of caspases offered by Reaction Biology Corporation. This data confirmed the potent inhibitory capacity of 2b versus caspase 1 (IC50 = 0.204nM); however, the IC50 values found versus caspase 4 (IC50 = 14.5nM) and caspase 8 (IC50 = 3. nM) differed slightly from the reported Ki values. The results against caspase 6 (IC50 ≥ 10,000nM) and caspase 9 (IC50 = 5.07nM) were significantly different from those reported by Randle and coworkers [7]. The Reaction Biology Corporation panel also included caspase 5 (IC50 = 10.6nM), caspase 10 (IC50 = 66.5nM) and caspase 14 (IC50 = 58.5nM), and the data presented here represents the first disclosure of the IC50 values for 2b versus these targets. The results for 4 demonstrated an impressive potency against caspase 1 (IC50 = 0.023nM) and a similar selectivity profile as 2b. The only prominent divergence between the selectivity profiles of 4 and 2b was a sharp drop in the ability to inhibit caspase 14 (IC50 = 801nM and IC50 = 58.5nM, respectively). The caspase 1 inhibition data generated in this panel for 3 and 16 was similar to the data generated in our caspase 1 assay, with reported IC50 values of 43.4 nM and 2.58 nM, respectively. A particularly interesting aspect of these molecules was the high selectivity for caspase 1. NCGC-00183681 (16) registered an IC50 value of 91.5nM versus caspase 9. All other activities were above the 1μM threshold. In addition to the primary molecules of this study, we were interested in establishing cyanopropanoates as general caspase directing ‘warheads’ for future utility in the search for other potent and selective small molecule inhibitors of caspases. As such, we included the general nitrile-Asp directing group into the common peptide caspase inhibitors YVAD. The resulting agent, YVAD-CN (20), was profiled, and the results clearly demonstrate that cyanopropanoates represent a general moiety for reversible, covalent modification of caspases. Based upon this data, we conclude that these agents do not represent promiscuous covalent protein modifiers.
The acid moiety can cause issues with membrane permeability (though not always). In order to combat this, we have provided a pro-drug version of NCGC-00183434/CID-44620939.
3.4. SAR Tables
Table 1Investigation of SAR around aryl ring
 | |||||
|---|---|---|---|---|---|
| Entry | Ar | Internal IDa | CID | SID | IC50 (nM)b |
| 1 |
 | NCGC-00183434 | 44620939 | 87544173 | 0.47 |
| 2 |
 | NCGC-00241070 | 46916205 | 99380812 | 0.424 |
| 3 |
 | NCGC-00241069 | 46916204 | 99380811 | 2.09 |
| 4 |
 | NCGC-00241073 | 46916207 | 99380814 | 0.44 |
| 5 |
 | NCGC-00241071 | 46916206 | 99380813 | 5.36 |
- a
All compounds synthesized at NCGC
- b
IC50 values were determined at NCGC utilizing a fluorescent enzyme assay using the appropriate AFC-labeled peptide.
Table 2IC50 values for selected compounds versus caspase panel
| Compound | Caspase 1 (nM) | Caspase 3 (nM) | Caspase 4 (nM) | Caspase 5 (nM) | Caspase 6 (nM) | Caspase 7 (nM) | Caspase 8 (nM) | Caspase 9 (nM) | Caspase 10 (nM) | Caspase 14 (nM) |
|---|---|---|---|---|---|---|---|---|---|---|
| VRT-043198(2b) (Drug) | 0.204 | >10000 | 14.5 | 10.6 | >10000 | >10000 | 3.3 | 5.07 | 66.5 | 58.5 |
| 3 (Nitrile ester) | 43.4 | >10000 | >10000 | 1570 | >10000 | >10000 | >10000 | 1610 | >10000 | >10000 |
| 4 (Nitrile acid) | 0.023 | >10000 | 13.8 | 3.60 | >10000 | >10000 | 25.2 | 2.17 | 89.7 | 801 |
| 16 (Nitrile tetrazole) | 2.58 | >10000 | 1380 | 1300 | >10000 | >10000 | >10000 | 91.5 | >10000 | >10000 |
| 20 YVAD-CN | 2.16 | >10000 | 114 | 29.0 | >10000 | >10000 | 726 | 297 | 187 | 116 |
| Ac-LEHD-CHO (standard) | 15.0 | ND | 81.7 | 21.3 | ND | ND | 3.82 | 49.2 | 40.4 | 134 |
| Ac-DEVD-CHO (standard) | ND | 3.04 | ND | ND | 122 | 3.54 | ND | ND | ND | ND |
- a
Data was generated by Reaction Biology (http://www
.reactionbiology.com/). Data is presented as an IC50’s using a (Z-LEHD)2-R110 tetrapeptide substrate for caspase 1, 4, 5, 8, 9, 10, 14 and a (Z-DEVD)2-R110 tetrapeptide substrate for caspase 3, 6 and 7. Data represents the results from three separate experiments.
3.5. Cellular Activity
Cellular studies have not yet been run with these agents.
3.6. Profiling Assays
We have profiled and selected analogs for absorption, distribution, metabolism and elimination (ADME) properties for chosen compounds. As such, 1, 2b, 3, 4 and 16 were submitted to Cyprotex for a profile of bi-directional Caco-2 permeability, plasma protein binding (both human and rat) and microsomal stability (both human and rat) studies (Table 3). All agents possessed relatively low A to B permeability; however, the pro-drug 1 and the ester 3 had moderately better levels. The high B to A levels reported for 1 and 3 strongly suggested an active transport mechanism, and a control experiment with verapamil confirmed that these agents are substrates for Pgp efflux. Unsurprisingly, the free acids 2b and 4 and the tetrazole 16 had significantly higher free fractions in both human and rat protein binding assays relative to the more hydrophobic prodrug 1 and ethyl ester 3. The clearance rates (Clint) and t1/2 for 2b, 3, 4 and 16 were all moderate. The ester 3 was noted to possess a slight degree of degradation in liver microsomes without NADPH as a cofactor, suggesting a non-enzymatic related degradation mechanism. The pro-drug 1 possessed minimal ability to be metabolized by liver microsomes and a t1/2 of >9400 minutes. It is unknown how this extended stability affects this agent’s toxicity profile.
Table 3
In vitro ADME properties for selected compounds.
4. Discussion
A few areas of SAR were explored around NCGC-00183434/CID-44620939/ML132. The first investigation was to look at replacement of the 3-chloro-4-anilino-benzene ring with more hydrophobic groups. Replacement of the p-aniline with a p-methoxy or switching to the 3,5-dichlorobenzene resulted in no significant change in activity (compare entries 1, 2 and 4 in Table 1). O-substitution resulted in a 5 fold loss in activity (entry 3), while the p-trifluoromethylbenzene analog saw a 10-fold loss in potency. Investigation around the warhead was undertaken and an ethyl ester prodrug was made vide supra as well as a tetrazole version, which is an established carboxylic acid mimetic. As can be seen in Table 2, the tetrazole analog had lower activity, but interestingly was slightly more selective than the acid version. As expected, the ester prodrug version (NCGC-00185682) did have slightly better caco-2 permeability vide supra (Table 3).
4.1. Comparison to existing art and how the new probe is an improvement
NCGC-00183434/CID-44620939/ML132 is the most potent caspase 1 inhibitor reported to date. It also possesses a unique selectivity pattern relative to other reported caspase inhibitors.
4.2. Mechanism of Action Studies
We examined the binding mechanism of these agents through molecular modeling. Several crystal structures of caspase 1 exist, including structures with reversible and non-reversible inhibitors (PDB codes: 1BMQ, 1IBC, 1ICE, 1RWK, 1RWM, 1RWN, 1RWO, 1RWP, 1RWV, 1RWW, 1RWX, 1SC1, 1SC3, 1SC4, 2FQQ, 2H48, 2HBQ, 2HBR, 2HBY, 2HBZ, 2FQR, 2FQS, 2FQU, 2FQZ) [8–11]. We identified 2HBQ as the best template for 4 (2HBQ is a co-crystal of caspase 1 and Z-VAD-FMK). We applied the presumption of a covalent reversible mechanism of inhibition when building a model for binding of 4. The nitrile carbon was therefore held at a proximal distance (2.6 Å) from the catalytic cysteine residue (C285) by constraint docking, and flexibility was granted to the remainder of the small molecule to achieve an optimal binding pose using FRED [12]. The results are shown in figure 4 and demonstrate complementarity between the peptidic fragment of 4 and the peptide binding domain of caspase 1. Key interactions were noted for the acid moiety and arginine residues 341 and 179 in similar fashion to other Asp containing small molecule caspase 1 inhibitors. While direct interrogation of a covalent interaction between the nitrile and C285 was not pursued in our model, this representation does illustrate the open binding cavity that accommodates the tetrahedral intermediate that forms as a result of covalent binding with aldehyde based inhibitors (a mimetic of the hemithiolacetal intermediate associated with transition state 1 (TS1) during proteolysis). In contrast, covalent interactions between a thiol and a nitrile form a thioimidate intermediate that mimics transition state 2 (TS2) of an enzymatic proteolytic event between a cysteine proteases and a substrate. Ménard and coworkers examined aldehyde and nitrile inhibitors of papain and found that the thioimidate intermediate engages the oxyanion hole interaction in a manner that more closely mimics the natural process of hydrolysis during proteolysis [13]. This may have consequences for both the binding affinity of nitrile-based cysteine proteases inhibitors and their ultimate resolution through hydrolysis of the thioimidate intermediate.
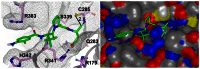
Figure 4
Molecular model (ribbon and space filling) of NCGC-00183434/CID-44620939/ML132 (4) bound to caspase 1.
4.3. Planned Future Studies
Recently, the Petsko and Ringe labs have identified the human enzyme caspase-1 as the protease that cleaves α-synuclein in vivo, producing a C-terminal truncated fragment that nucleates the aggregation of αS. Synucleinopathies are a class of neurologic disorders that together afflict millions of Americans. They are characterized by neurocytoplasmic inclusions containing primarily α-synuclein, an abundant neuronal protein whose cellular functions are poorly understood. The most prevalent synucleinopathy is Parkinson’s disease, a devastating neurodegenerative condition that kills dopaminergic neurons, leading to tremor, slowness of movement, and eventual paralysis and death. We plan to continue our optimization of the existing probe compound in a manner that will allow it to penetrate the blood-brain barrier, thus allowing the Wells and Petsko labs to examine the role of caspase 1 in synucleinopathies, including Parkinson’s disease.
Probe properties
Properties Computed from Structure
| Calculated Property | Probe Identity |
|---|---|
| CID_44620939 (MLS003178557) | |
| Molecular Weight [g/mol] | 477.94122 |
| Molecular Formula | C22H28ClN5O5 |
| XLogP3-AA | 1.6 |
| H-Bond Donor | 4 |
| H-Bond Acceptor | 7 |
| Rotatable Bond Count | 8 |
| Tautomer Count | 10 |
| Exact Mass | 477.177897 |
| MonoIsotopic Mass | 477.177897 |
| Topological Polar Surface Area | 166 |
| Heavy Atom Count | 33 |
| Formal Charge | 0 |
| Isotope Atom Count | 0 |
| Defined Atom StereoCenter Count | 3 |
| Undefined Atom StereoCenter Count | 0 |
| Defined Bond StereoCenter Count | 0 |
| Undefined Bond StereoCenter Count | 0 |
| Covalently-Bonded Unit Count | 1 |
| Complexity | 820 |
5. References
- 1.
- Martinon F, Tschopp J. Cell. 2004;117:561–574. [PubMed: 15163405]
- 2.
- Braddock M, Quinn A. Nat. Rev Drug Discovery. 2004;3:1–10. [PubMed: 15060528]
- 3.
- Linton SD. Curr. Top. Med. Chem. 2005;5:1697–1717. [PubMed: 16375749]
- 4.
- Falgueyret J-P, Oballa RM, Okamoto O, Wesolowski G, Aubin Y, Rydzewski RM, Prasit P, Riendeau D, Rodan SB, Percival MD. J. Med. Chem. 2001;44:94–104. [PubMed: 11141092]
- 5.
- Mallari JP, Shelat AA, Obrien T, Caffrey CR, Kosinski A, Connelly M, Harbut M, Greenbaum D, McKerrow JH, Guy RK. J. Med. Chem. 2008;51:545–552. [PubMed: 18173229]
- 6.
- Mott BT, Ferreira R, Simeonov A, Jadhav A, Ang K-H, Leister W, Shen M, Silveira JT, McKerrow JH, Inglese J, Austin CP, Thomas CJ, Shoichet BK, Maloney DJ. J. Med. Chem. 2010;53:52–60. [PMC free article: PMC2804034] [PubMed: 19908842]
- 7.
- Wannamaker W, Davies R, Namchuk M, Pollard J, Ford P, Ku G, Decker C, Charifson P, Weber P, Germann UA, Kuida K, Randle JCR. J. Pharmacol. Exp. Ther. 2007;321:509–516. [PubMed: 17289835]
- 8.
- Wilson KP, Black J-AF, Thomason JA, Kim EE, Griffith JP, Navia MA, Murcko MA, Chambers SP, Aldape RA, Raybuck SA, Libingston DJ. Nature. 1994;370:270–275. [PubMed: 8035875]
- 9.
- Okamoto Y, Anan H, Nakai E, Morihira K, Yonetoku Y, Kurihara H, Sakashita H, Terai Y, Takeuchi M, Shibanuma T, Isomura Y. Chem. Pharm. Bull. 1999;47:11–21. [PubMed: 9987822]
- 10.
- Romanowski MJ, Scheer JM, O’Brien T, McDowell RS. Structure. 2004;12:1361–1371. [PubMed: 15296730]
- 11.
- Scheer JM, Romanowski MJ, Wells JA. Proc. Nat. Acad. Sci. U. S. A. 2006;103:7595–7600. [PMC free article: PMC1458511] [PubMed: 16682620]
- 12.
- Santa Fe, NM: OpenEye Scientific Software, Inc.; http://www
.eyesopen.com/ - 13.
- Dufour E, Storer AC, Ménard R. Biochemistry. 1995;34:9136–9143. [PubMed: 7619812]
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- A highly potent and selective caspase 1 inhibitor that utilizes a key 3-cyanopropanoic acid moiety.[ChemMedChem. 2010]A highly potent and selective caspase 1 inhibitor that utilizes a key 3-cyanopropanoic acid moiety.Boxer MB, Quinn AM, Shen M, Jadhav A, Leister W, Simeonov A, Auld DS, Thomas CJ. ChemMedChem. 2010 May 3; 5(5):730-8.
- Structure-based discovery of a novel non-peptidic small molecular inhibitor of caspase-3.[Bioorg Med Chem. 2008]Structure-based discovery of a novel non-peptidic small molecular inhibitor of caspase-3.Sakai J, Yoshimori A, Nose Y, Mizoroki A, Okita N, Takasawa R, Tanuma S. Bioorg Med Chem. 2008 May 1; 16(9):4854-9. Epub 2008 Mar 21.
- Discovery of indole tetrafluorophenoxymethylketone-based potent novel small molecule inhibitors of caspase-3.[Org Med Chem Lett. 2012]Discovery of indole tetrafluorophenoxymethylketone-based potent novel small molecule inhibitors of caspase-3.Samiulla DS, Naidu A, Rao GV, Ramachandra M. Org Med Chem Lett. 2012 Jul 16; 2(1):27. Epub 2012 Jul 16.
- Review Thiol-dependent enzymes and their inhibitors: a review.[Curr Med Chem. 2002]Review Thiol-dependent enzymes and their inhibitors: a review.Leung-Toung R, Li W, Tam TF, Karimian K. Curr Med Chem. 2002 May; 9(9):979-1002.
- Review Development of nitrile-based peptidic inhibitors of cysteine cathepsins.[Curr Top Med Chem. 2010]Review Development of nitrile-based peptidic inhibitors of cysteine cathepsins.Frizler M, Stirnberg M, Sisay MT, Gütschow M. Curr Top Med Chem. 2010; 10(3):294-322.
- A small molecule inhibitor of Caspase 1 - Probe Reports from the NIH Molecular L...A small molecule inhibitor of Caspase 1 - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...