

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

The specific aim of this project is to identify, via a functional high-throughput screening (HTS) approach, small molecule positive allosteric modulators (PAMs) and/or allosteric agonists of the M1 muscarinic acetylcholine receptor (mAChR) that are cell permeable, possess submicromolar potency, and show greater than 10-fold selectivity over the other mAChRs (M2-M5). The currently identified probe ML169 (CID-44475955) can be used for in vitro molecular pharmacology and electrophysiology experiments to study the receptor trafficking profile, and the role of selective M1 receptor activation by this unique M1 PAM chemotype. Use of this probe alongside the authors' initially identified M1 PAM probe, ML137 (CID-44251556) could improve understanding of the M1 signaling pathway and elucidate the difference, if any, between high and low ACh fold-shift compounds due to their different pharmacological characteristics. This probe possesses high selectivity versus M2-M5, as well as a large panel of GPCRs, ion channels and transporters. While in vivo studies are possible, it has not been investigated for such uses. Probes developed from these efforts will greatly advance the current state of the art by aiding in the understanding of M1's role in cell-based physiology, and may extend the clinical understanding of psychotic and cognitive symptoms associated with neurodegenerative disorders like Alzheimer's Disease and schizophrenia.
Assigned Assay Grant #: MH077606-01
Screening Center Name & PI: Vanderbilt Screening Center for GPCRs, Ion Channels and Transporters, C. David Weaver
Chemistry Center Name & PI: Vanderbilt Specialized Chemistry Center for Accelerated Probe Development, Craig W. Lindsley
Assay Submitter & Institution: P. Jeffrey Conn, Vanderbilt University
PubChem Summary Bioassay Identifier (AID): AID-2543
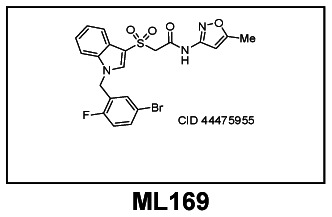
Probe Structure & Characteristics
2-((1-(5-bromo-2-fluorobenzyl)-1H-indol-3-yl)sulfonyl)- N-(5-methylisoxazol-3-yl)acetamide
MW = 506.4, logP = 3.9, TPSA = 94.2 Å2
| CID/ML# | Target Name | IC50/EC50 (nM) [SID, AID] | Anti-target Name(s) | IC50/EC50 (μM) [SID, AID] | Selectivity | Secondary Assay(s) Name: IC50/EC50 (nM) [SID, AID] |
|---|---|---|---|---|---|---|
| CID-44475955 ML169 | M1 | 1380 [SID-85756541,AID-2651] | M2, M3, M4, M5 Ricerca Lead Profiling | > 30 μM [SID-85756541, AID-2430, AID-2428, AID-2438, AID-2433] | >30 | ACh Fold-shift (45-fold) [SID-85756541, AID-2434] |
Recommendations for the scientific use of this probe
This probe (CID 44475955) can be used for in vitro molecular pharmacology and electrophysiology experiments to study the receptor trafficking profile and the role of selective M1 receptor activation by this unique M1 PAM chemotype. Use of this probe alongside our initial M1 PAM probe (CID 44251556) could improve our understanding of the M1 signaling pathway and elucidate the difference, if any, between high and low ACh fold-shift compounds, due to their different pharmacological characteristics. This probe possesses high selectivity versus M2–M5, as well as a large panel of GPCRs, ion channels and transporters. While in vivo studies are possible, it has not been investigated for such uses.
Specific AIM
To identify small molecule positive allosteric modulators (PAMs) and/or allosteric agonists of the M1 muscarinic acetylcholine receptor that are cell permeable, possess submicromolar potency and show greater than 10-fold selectivity over the other mAChRs (M2–M5) employing a functional HTS approach. Out of this effort aimed at M1, which afforded a highly selective M1 antagonist (CID 24768606), a highly selective M1 allosteric agonist (CID 25010775) and a highly selective M1 PAM (CID 44251556), we also identified and optimized the first M5 ligand, an M5 PAM (CID 42633508). Starting from an unrelated lead, belonging to a novel chemotype, we have now been able to develop a second highly selective M1 PAM with a low-micromolar EC50 and a greatly improved ACh fold-shift. Another MLSCN screening effort identified a highly selective M4 PAM (CID 864492); thus, two MLSCN/MLPCN screens have provided a toolkit of highly selective mAChR ligands available from the MLPCN to study individual mAChR function both in vitro and in vivo.
Significance
The five cloned muscarinic acetylcholine receptor subtypes (mAChR1-5 or M1–M5) are known to play highly important and diverse roles in many basic physiological processes.1–3 Correspondingly, muscarinic agonists and antagonists targeting one or more subtypes have been used preclinically and clinically for research and treatment of a wide range of pathologies.3,4 Based on the high sequence homology of the mAChRs across subtypes, and particularly within the orthosteric acetylcholine (ACh) binding site, discovery of truly subtype-selective compounds has proven historically difficult. Due to the scarcity of selective compounds, a detailed understanding of the precise roles of each subtype in neurobiology and in various central nervous system (CNS) disorders has thus remained elusive.3,4 In numerous Phase II and III clinical trials, pan-mAChR agonists were shown to improve cognitive performance in AD patients, but the GI-and/or cardiovascular side effects, resulting from activation of peripheral mAChRs, were deemed intolerable and the trials were discontinued.5,6 Importantly, several pan-mAChR agonists demonstrated a decline in the concentration of Aβ42 in the cerebral spinal fluid of AD patients, suggesting that mAChR activation has the potential to be disease modifying as well as providing palliative cognitive therapy.7 More recent studies in 3xTg-AD mice further support a disease modifying role for mAChR activation, and several Ph III trials demonstrated that mAChR activation lowered Aβ42 in patients.8 Interestingly, the M1/M4 preferring xanomeline, in addition to improving cognitive performance, had robust therapeutic effects on the psychotic symptoms and behavioral disturbances associated with AD and recently published clinical trial data indicates efficacy in schizophrenic patients.9,10 Probes developed from these efforts will greatly advance the current state of the art by aiding in the understanding of M1’s role in cell-based physiology and may extend the clinical understanding of psychotic and cognitive symptoms associated with neurodegenerative disorders like Alzheimer’s Disease and schizophrenia.
Rationale
In recent years, major advances have been made in the discovery of highly selective agonists of other G protein-coupled receptors (GPCRs) that act at an allosteric site rather than the orthosteric binding site.11,12 By screening for compounds that act at an allosteric site on the receptor, it is anticipated that compounds that selectively activate M1 versus the other muscarinic subtypes may be identified.13–18 While allosteric M1 agonists have been identified, AC-42 and TBPB, they both suffer from undesirable ancillary pharmacology, poor physicochemical properties, poor pharmacokinetics and/or limited CNS exposure.19,20 Thus, to truly enable the biomedical community to dissect the relative contributions of selective M1 activation in preclinical models of AD and schizophrenia and to understand the role of M1 in the pronounced efficacy of the M1/M4 preferring xanomeline, improved M1 probes are required. Recently, a number of novel highly subtype-selective allosteric ligands for M1 and M4 have emerged from functional cell-based screening efforts – several are MLPCN probes along with the prototypical M1 PAM, BQCA.13–18,20–22 Although considerable interest was initially generated around BQCA, this level of interest appears to have waned and may point to a fatal flaw in its general chemotype (See Figure 2). Our initial report on the discovery of CID 3008304, a pan Gq M1, M3, M5 PAM, also described three other series of weak M1 PAMs, and established that different M1 PAM chemotypes displayed different modes of activity on downstream receptor signaling/trafficking despite similar profiles in Ca2+ assays.13 Thus, all allosteric M1 activation is not equivalent, and additional tool compounds representing diverse chemotypes are required to truly dissect and study M1 function in the CNS. Subsequent to our development of an M5 selective PAM from a pan Gq M1, M3, M5 PAM,21,22 we next optimized CID 3008304 for M1 PAM activity resulting in the first highly selective M1 PAM MLCPN probe (CID 44251556). For the above reasons associated with downstream receptor signaling/trafficking, we were simultaneously pursuing alternative leads in an attempt to add unique chemotypes to our tool kit of selective M1 activators.23
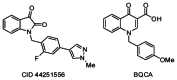
Figure 2
M1 PAMs, CID 44251556 and BQCA.
Assay Implementation and Screening
PubChem Bioassay Name
Discovery of Novel Allosteric Modulators of the M1 Muscarinic Receptor: Positive Allosteric Modulator (PAM)
List of PubChem bioassay identifiers generated for this screening project (AIDs)
AID-2651, AID-2425, AID-2428, AID-2430, AID-2434, AID-2433, AID-2438, AID-2626, and AID-2543.
PubChem Primary Assay Description
Chinese hamster ovary (CHO K1) cells stably expressing rat (r)M1 were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and cultured according to their recommendations. CHO cells stably expressing human (h) M2, hM3, and hM5 were generously provided by A. Levey (Emory University, Atlanta, GA); rM4 cDNA provided by T. I. Bonner (National Institutes of Health, Bethesda, MD) was used to stably transfect CHO-K1 cells purchased from the ATCC using Lipofectamine 2000. To make stable hM2 and rM4 cell lines for use in calcium mobilization assays, cell lines were cotransfected with a chimeric G protein (Gqi5) using Lipofectamine 2000. hM2, hM3, and hM5 cells were grown in Ham’s F-12 medium containing 10% heat-inactivated fetal bovine serum, 2 mM GlutaMax I, 20 mM HEPES, and 50 μg/mL G418 sulfate. hM2-Gqi5 cells were grown in the same medium supplemented with 500 μg/mL hygromycin B. Stable rM4 cells were grown in Dulbecco’s modified Eagle’s medium containing 10% heat-inactivated fetal bovine serum, 2 mM GlutaMax I, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 20 mM HEPES, and 400 μg/mL G418 sulfate; rM4-Gqi5 cells were grown in the same medium supplemented with 500 μg/mL hygromycin B. CHO cells stably expressing rM1, hM3, or hM5 were plated at a seeding density of 50,000 cells/100 μL/well. CHO cells stably coexpressing hM2/Gqi5 and rM4/Gqi5 were plated at a seeding density of 60,000 cells/100 μL/well. For calcium mobilization, cells were incubated in antibiotic-free medium overnight at 37 °C/5% CO2 and assayed the next day.
Calcium Mobilization Assay
Cells were loaded with calcium indicator dye [2 μM Fluo-4 acetoxymethyl ester (50 μL/well) prepared as a stock in DMSO and mixed in a 1:1 ratio with 10% Pluronic acid F-127 in assay buffer (1xHanks’ balanced salt solution supplemented with 20 mM HEPES and 2.5 mM probenecid, pH 7.4)] for 45 min at 37 °C. Dye was removed and replaced with the appropriate volume of assay buffer. All compounds were serially diluted in assay buffer for a final 2x stock in 0.6% DMSO. This stock was then added to the assay plate for a final DMSO concentration of 0.3%. Acetylcholine (EC20 concentration or full dose-response curve) was prepared at a 10x stock solution in assay buffer before addition to assay plates. Calcium mobilization was measured at 25 °C using a FLEXstation II (Molecular Devices, Sunnyvale, CA). Cells were preincubated with test compound (or vehicle) for 1.5 min before the addition of the agonist, acetylcholine. Cells were then stimulated for 50 s with a submaximal concentration (EC20) or a full dose-response curve of acetylcholine. The signal amplitude was first normalized to baseline and then as a percentage of the maximal response to acetylcholine.
Summary of Screen
This screen was performed in the pilot phase, the MLSCN, when the MLSMR compound collection at Vanderbilt only contained ~65,000 compounds. Results from the primary M1 screen of these compounds identified ~12 putative M1 PAMs with an average Z’ score of 0.70±0.09. The confirmation screen (singles at 10 μM) produced two lead compounds, one of which was optimized into our first M1 PAM probe (CID 44251556). The other, CID 2157678, represented a viable lead structure already endowed with good receptor subtype selectivity across the M3/M5 receptors (Figure 1). However, its potency upon reconfirmation from dry solid (M1 EC50 = 12.9 μM) was not particularly attractive and so a program was initiated to develop this lead into a pharmacologically more useful tool by improving its potency while maintaining its M2–M5 receptor subtype selectivity. Ultimately, it was envisioned that an M1 PAM not structurally related to the known M1 PAMs (CID 44251556 and BQCA, Figure 2) may possess a distinct pharmacology at the M1 receptor, thereby enabling an expanded understanding of the M1 muscarinic receptor’s downstream signaling/trafficking.

Figure 1
CRCs at M1–M5 for HTS lead CID 2157678.
Probe Chemical Lead Optimization Strategy
Our initial optimization strategy is outlined in Figure 3, and as SAR with allosteric ligands is often shallow, we employed an iterative parallel synthesis approach, along with targeted syntheses for structures encompassing more speculative modifications. Attempted modifications of the Eastern oxazole-amide, although not extensive, met with no success, returning only compounds with undetectable activity (Such as CID 44129586, 44634499, 44634501 and 44634503). In a straightforward attempt to reduce molecular weight the benzyl group attached to the indole nitrogen was omitted, but met with a similar lack of success (CID 751482, EC50 > 10 μM).
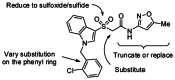
Figure 3
Initial optimization strategy for CID 2157678.
Accordingly, libraries were concomitantly prepared as shown in Scheme 1 and predominately surveyed diversity on the Southern benzyl moiety. Methyl thioglycolate (1) was arylated with indole using iodine and potassium iodide, followed by saponification of the ester using lithium hydroxide to give acid 2. This acid was then peptide coupled with 3-amino-5-methyl-isoxazole (3) employing PyClU (chlorodipyrrolidinocarbenium hexafluorophosphate), in DCE, with microwave heating to yield thioether 4. The sulfur was then oxidized to the sulfone with Oxone, thereby allowing the benzylation of the indole nitrogen to be performed as the final step in the preparation of the initial library (Analogs 5, Table 1). As would be expected for this benzylation, bis-alkylation was observed to a small extent, and occasionally allowed for the isolation of analogs like CID 44129599 (Figure 4). While it was not terribly surprising that these analogs were devoid of M1 PAM activity, it would eventually be ascertained that even the introduction of a solitary methyl group α to the amide abolished any measureable M1 PAM activity (CID 44634500, M1 EC50 > 10 μM, is the μ-methylated analog of the probe compound). Alternatively, if the Oxone oxidation step is omitted or replaced with a milder oxidant (e.g. FeCl3/H5IO6) then thioethers (CID 3305286) or sulfoxides (CID 44247543) analogous to the lead structure were produced. However only the sulfoxide (CID 44247543) retained measureable activity (M1 EC50 = 5.23 μM), but did not represent a significant improvement over its sulfone analog (5b, CID 44129591, Table 1).
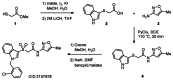
Scheme 1
Synthesis of CID 2157678 and the route to its analogs.
Table 1
Structures and activities of analogs 5.

Figure 4
A representative over-alkylation product from Scheme 1.
Focusing on the library of analogs appearing in Table 1, we quickly discovered that substitution in the 3-position of the phenyl ring was preferred. Moving the chlorine from the 2-position in the lead structure (5a) to the 3-position (5b) improved the M1 EC50 by about 50%. While, either a methoxy (5e) or a fluorine (5f) at this position afforded a small improvement in their EC50 values, a 2-fold increase in potency could be obtained by introducing a bromine at the 3-position providing 5h, which possessed an EC50 = 3.79 μM. The remainder of Table 1, servers to illustrated how steep the SAR was for this class of allosteric modulators. For example, the addition of a second halogen in examples 5k and 5l eliminated the measurable efficacy displayed by their mono-halogenated analogs (5h and 5b, respectively). In general it could also be concluded that substitution at the 4-position on these aryl groups was uniformly detrimental. Having learned from the initial library that proper substitution at the 3-position of the benzyl group could be beneficial, and with the bromide 5h in hand we sought to introduce various aryl rings at this location. The small library of analogs 6 appearing in Table 2 were prepared via microwave-assisted Suzuki couplings between 5h and the requisite boronic acid. Impetus for the introduction of the pyrazole moiety found in 6a–c, came from its successful incorporation into our previous M1 PAM probe (CID 44251556) and BQCA analogs (Figure 2). Gratifyingly, the N-methyl pyrazole 6a did have measureable activity but did not impart a significant improvement in potency over the corresponding bromide (5h). Divergent from the SAR established during the development of CID 44251556, the unalkylated pyrazole 6b was now preferred, possessing an M1 EC50 = 2.19 μM. In contrast to the N-methyl pyrazole 6a, the larger sec-butyl substituent of 6c was not tolerated, and neither were the other heterocycles of the remaining analogs (6d–h).
Table 2
Structures and activities of analogs 6.
To further the development of this novel series of M1 PAMs we focused on three of the more potent analogs (5b, 5h and 6b) and applied a fine-tuning process of introducing fluorine atoms at various locations to provide analogs 7 (Table 3). Across the series, substitution at the 4-position was uniformly not tolerated (7a–c), consistent with the SAR appearing in Table 1. Bis-fluorination of the indole ring eroded activity in the context of the bromine analog 7d but conversely augmented the activity of the pyrazole congener 7e. This type of subtle/confounding SAR was similarly observed with respect to fluorination at the R2 position in analogs 7f–h. While the presence of a fluorine at R2 was neutral or slightly beneficial in the context of the chlorine analog (7f), its presence resulted in clearly decreased activity for both the bromine and pyrazole analogs, 7g and 7h. Lastly, the introduction of a single fluorine at the R6 position could either be moderately detrimental in the context of pyrazole 7i or decidedly beneficial with respect to bromine analog 7j, where an almost 3-fold improvement in potency was observed. In this manner, both CID 44475955 (7j) and the related difluorindole analog CID 44475948 (7e) were developed and chosen for further evaluation (For clarity, these compounds are shown in Figure 5).
Table 3
Structures and activities of analogs 7.

Figure 5
The two most potent M1 PAMs developed from the lead CID 2157678.
Both CID 44475955 and CID 44475948 were highly selective PAMs for the M1 receptor, displaying minimal/no potentiation of the M2–M5 receptors up to 30 μM (Figure 6A–B). Similarly, both compounds demonstrated impressive left-ward shifts in the potency of ACh in M1 ACh Concentration Response Curve (CRC) fold-shift experiments (Figure 6C). When tested at 30 μM, the bromine analog 7j engendered a 45-fold increase in ACh activity, while the pyrazole analog 7e increased ACh activity 98-fold. These levels of potentiation are very similar to those seen for BQCA14,15 and represent a sizable improvement over the earlier M1 PAM probe molecule CID 44251556 which produced only a 3-fold shift in an identical experiment. Still, these experiments did not reveal a definitive superiority for either molecule.

Figure 6
A) CRCs for CID 44475955 at M1–M5; B) CRCs for CID 44475948 at M1–M5; C) Fold-Shift for CID 44475955 and CID 44475948 at 30 μM.
An examination of calculated physical properties and practical considerations were more helpful in choosing the preferred probe. The in silico values for CID 44475955 and CID 44475948 appearing in Table 4 were calculated using TRIPOS software. Also included in Table 4 are the averages from the MDDR database of compounds both entering Phase I and launched drugs. Once again both compounds were very similar across a number of parameters. However, CID 44475955 possessed superior values for both total polar surface area (TPSA) and its number of hydrogen bond donors (Hdon). These two discrepancies both point to a lower probability of crossing the blood brain barrier for CID 44475948. Although not tested in vivo, this would lead to the prediction that CID 44475955 has a better chance of being a CNS penetrant molecule, and therefore a potentially superior MLPCN probe compound for the muscarinic receptors present in the CNS. Additionally, from a synthesis standpoint, the greater ease and lower cost for the acquisition of indole (Scheme 1) over 4,6-difluoroindole (the starting material for CID 44475948) makes CID 44475955 the more practical and cost-effective probe molecule.
Table 4
Calculated Property Comparison with MDDR Compounds.
To more fully characterize this novel M1 PAM, CID 44475995 was tested at Ricerca’s (formerly MDS Pharma’s) Lead Profiling Screen (binding assay panel of 68 GPCRs, ion channels and transporters screened at 10 μM), and was, thus far, found to not significantly interact with 16 out of the 16 assays conducted (Remaining 52 pending).24 Thus, CID 44475955 is highly selective and can be used to dissect the role of M1 in vitro. Furthermore, when used in conjunction with our earlier M1 PAM probe molecule, the potentially different effects caused by an ACh low fold-shift probe (CID 44251556, 3-fold) versus those of an ACh high fold-shift probe (CID 44475955, 45-fold) may reveal the importance of this parameter towards the treatment of different disease states.
In summary, from an initial MLSCN screening campaign of just 65,000 compounds a second, potent and highly selective M1 PAM (CID 44475955) has been identified which is structurally distinct from our initial M1 PAM probe (CID 44251556). CID 44475955, similar to our previous probe, possesses comparable potency to BQCA, but now in contrast to our first probe, also shares BQCA’s ability to produce a large left-ward shift in the activity of ACh at the M1 receptor. CID 44475955 now represents the third known chemotype to provide potent and selective M1 positive allosteric modulation. Further in vitro and in vivo characterization of CID 44475955 is in progress in the assay submitter’s lab, and data will be reported in due course.
Synthetic procedure and spectral data for CID 44475955

CID 44475955, 2-((1-(5-bromo-2-fluorobenzyl)-1H-indol-3-yl)sulfonyl)-N-(5-methylisoxazol-3-yl)acetamide [ML169]
To a solution of indole (3.00 g, 25.6 mmol) and methyl thioglycolate (2.40 mL, 25.6 mmol) in methanol:water (80 mL : 20 mL) was added iodine (6.50 g, 25.6 mmol) and potassium iodide (4.25 g, 25.6 mmol). The reaction mixture was stirred at ambient temperature for 60 hours. Methanol was removed in vacuo and the aqueous layer diluted with a saturated solution of sodium bicarbonate and extracted with ethyl acetate. The organic layer was dried over magnesium sulfate, evaporated in vacuo and the resulting residue was purified on a silica gel column (0–100% ethyl acetate:hexanes over 33 min) to afford compound 8 as an oil (LCMS >98%). Compound 8 was dissolved in a mixture of tetrahydrofuran (20 mL) and 2.0M aqueous LiOH (15 mL), then stirred vigorously at ambient temperature for 30 minutes. Tetrahydrofuran was removed in vacuo, the aqueous layer neutralized with 1.2 N HCl and extracted with CH2Cl2. The organic layer was dried over magnesium sulfate and removed in vacuo to produce an oily residue. Upon diluting the residue in dichloromethane a reddish-brown solid formed which was filtered and dried to yield compound 9 (2.00 grams, 9.65 mmol, 38% yield over 2 steps, LCMS >98%).
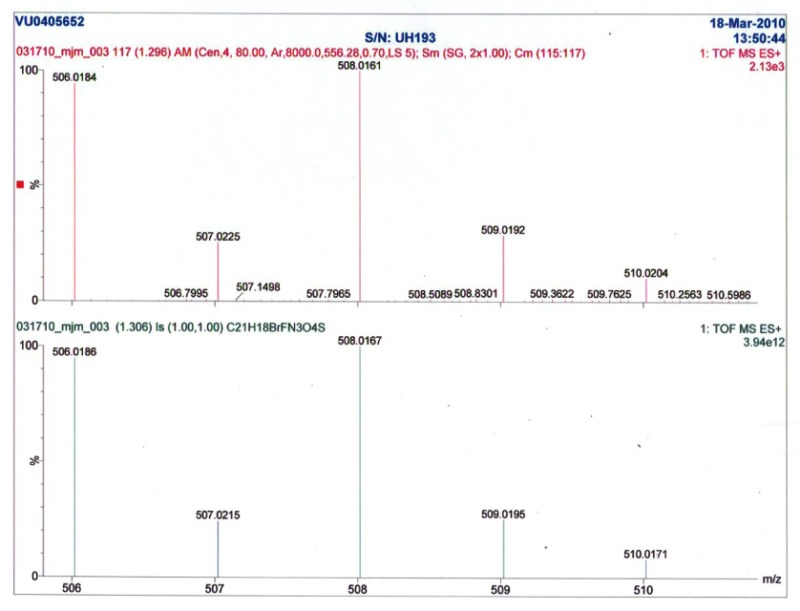
Compound 9 (650 mg, 3.14 mmol), 3-amino-5-methyl-isoxazole (616 mg, 6.28 mmol), PyClU (2.00 g, 6.28 mmol), and DIEA (1.36 mL, 7.85 mmol) were added to dichloroethane (25 mL) and microwave irradiated at 110 °C for 20 minutes. After cooling, the solvent was removed in vacuo and the remaining residue purified on a silica gel column (0–70% ethyl acetate:hexanes over 33 min) to yield compound 10 (642 mg, 2.23 mmol, 71% yield, LCMS >98%). Compound 10 (502 mg, 1.77 mmol) was dissolved in 25 mL (9:1, methanol:water) and Oxone (10.0 g, 17.7 mmol) was added. Stirring at ambient temperature continued overnight. Water (20 mL) was added and the mixture extracted with ethyl acetate (3x20 mL). The organics were combined, dried over magnesium sulfate, and concentrated in vacuo to give an oily residue which was purified on silica gel (0–50% ethyl acetate:hexanes over 19 min) to yield compound 11 (500 mg, 1.57 mmol, 88% yield, LCMS >98%). In a 5 mL microwave vial, compound 11 (55.0 mg, 0.174 mmol) was dissolved in DMF (3 mL) and cooled to 0 °C. Sodium hydride (60% by weight, 14.0 mg, 0.348 mmol) was then added in one portion and the reaction mixture vigorously stirred at 0 °C for 15 minutes. 4-bromo-2-bromomethyl-1-flouro-benzene (51.0 mg, 0.191 mmol) was added in one portion and the reaction mixture was stirred while being allowed to warm to ambient temperature over 3 hours. The reaction mixture was quenched with water (2 mL) and the solution was extracted with ethyl acetate (3x4 mL). The combined organics were dried over magnesium sulfate, concentrated in vacuo to give an oily residue which was purified on silica gel (0–70% ethyl acetate:hexanes over 19 min) to yield CID 44475955 (45 mg, 0.088 mmol, 51% yield). LCMS >98% 214 nm, RT = 1.34 min, m/z = 506 ([79Br]m+1), 508 ([81Br]m+1). 1H NMR (400 MHz, DMSO-d6) 11.27 (s, 1H), 8.23 (s, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.56–7.58 (m, 1H), 7.47 (dd, J = 2.4 Hz, 6.4 Hz, 1H), 7.35–7.23 (m, 3H), 6.54 (s, 1H), 5.62 (s, 2H), 4.43 (s, 2H), 2.37 (s, 3H), HRMS found: 506.0184; calculated for C21H17BrFN3O4S: 506.0185.
MLS#s
002700179 (Probe, 500 mg), 002700180, 002700181, 002700182, 002700183, 002700184
Bibliography
- 1.
- Bonner TI, Buckley NJ, Young AC, Brann MR. Science. 1987;237:527–532. [PubMed: 3037705]
- 2.
- Bonner TI, Young AC, Brann MR, Buckley NJ. Neuron. 1988;1:403–410. [PubMed: 3272174]
- 3.
- Wess J. Annu. Rev. Pharmacol. Toxicol. 2004;44:423–450. [PubMed: 14744253]
- 4.
- Langmead CJ, Watson J, Reavill C. Pharmacol. Ther. 2008;117:232–243. [PubMed: 18082893]
- 5.
- Eglen RM, Choppin A, Dillon MP, Hedge S. Curr. Opin. Chem. Biol. 1999;3:426–432. [PubMed: 10419852]
- 6.
- Felder CC, Bymaster FP, Ward J, DeLapp N. J.Med. Chem. 2000;43:4333–4353. [PubMed: 11087557]
- 7.
- Bodick NC, Ofen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, Shannon HE, Tollefson GD, Rasmussen K, Bymster FP, Hurley DJ, Potter WZ, Paul SM. Arch. Neurol. 1997;54:465–473. [PubMed: 9109749]
- 8.
- Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, LaFerla FM. Neuron. 2006;49:671–682. [PubMed: 16504943]
- 9.
- Fisher A. Neurodegener. Dis. 2008;5:237–240. [PubMed: 18322400]
- 10.
- Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Am. J. Psychiatry. 2008;165:1033–1039. [PubMed: 18593778]
- 11.
- Conn PJ, Christopoulos A, Lindsley CW. Nat. Rev. Durg Disc. 2009;8:41–54. [PMC free article: PMC2907734] [PubMed: 19116626]
- 12.
- Bridges TM, Lindsley CW. ACS Chem. Biol. 2008;3:530–542. [PubMed: 18652471]
- 13.
- Marlo JE, Niswender CM, Days EL, Bridges TM, Xiang Y, Rodriguez AL, Shirey JK, Brady AE, Nalywajko T, Luo Q, Austin CA, Williams MB, Kim K, Williams R, Orton D, Brown HA, Lindsley CW, Weaver CD, Conn PJ. Mol. Pharmacol. 2009;75:577–588. [PMC free article: PMC2684909] [PubMed: 19047481]
- 14.
- Ma L, Seager M, Wittman M, Bickel N, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danzinger A, Regan C, Garson S, Doran S, Kreatsoulas C, Veng L, Lindsley CW, Shipe W, Kuduk S, Jacobson M, Sur C, Kinney G, Seabrook GR, Ray WJ. Proc. Natl. Acad Sci. USA. 2009;106:15950–15955. [PMC free article: PMC2732705] [PubMed: 19717450]
- 15.
- Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Jadhav SB, Menon U, Christain EP, Doherty JJ, Quirk MC, Snyder DH, Levey AI, Watson ML, Nicolle MM, Lindsley CW, Conn PJ. J. Neurosci. 2009;29:14271–14286. [PMC free article: PMC2811323] [PubMed: 19906975]
- 16.
- Yang FV, Shipe WD, Bunda JL, Nolt MB, Wisnoski DD, Zhao Z, Barrow JC, Ray WJ, Ma L, Wittman M, Seager M, Koeplinger K, Hartman GD, Lindsley CW. Bioorg. Med. Chem. Lett. 2010;20:531–536. [PubMed: 20004574]
- 17.
- Lebois EP, Bridges TM, Dawson ES, Kennedy Jp, Xiang Z, Jadhav SB, Yin H, Meiler J, Jones CK, Conn PJ, Weaver CD, Lindsley CW. ACS Chemical Neurosci. 2010;1:104–121. [PMC free article: PMC3180826] [PubMed: 21961051]
- 18.
- Spalding TA, Trotter C, Skajaerbaek N, Messier TL, Currier EA, Burstein ES, Li D, Hacksell U, Brann MR. Mol. Pharm. 2002;61:1297–1302. [PubMed: 12021390]
- 19.
- Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane AS, Bridges TM, Kennedy JP, Bradley SR, Peterson TE, Ansari MW, Baldwin RM, Kessler RM, Deutch AY, Lah JJ, Levey AI, Lindsley CW, Conn PJ. J. Neurosci. 2008;28:10422–10433. [PMC free article: PMC2577155] [PubMed: 18842902]
- 20.
- Brady A, Jones CK, Bridges TM, Kennedy PJ, Thompson AD, Breininger ML, Gentry PR, Yin H, Jadhav SB, Shirey J, Conn PJ, Lindsley CW. J. Pharm. & Exp. Ther. 2008;327:941–953. [PMC free article: PMC2745822] [PubMed: 18772318]
- 21.
- Bridges TM, Marlo JE, Niswender CM, Jones JK, Jadhav SB, Gentry PR, Weaver CD, Conn PJ, Lindsley CW. J. Med. Chem. 2009;52:3445–3448. [PMC free article: PMC3875304] [PubMed: 19438238]
- 22.
- Bridges TM, Kennedy JP, Cho HP, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2010;20:558–562. [PMC free article: PMC3177601] [PubMed: 20004578]
- 23.
- Bridges TM, Kennedy JP, Cho HP, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. in press.
- 24.
- For information on the Ricerca (formerly MDS Pharma) Lead Profiling Screen see: https:
//pharmacology .ricerca.com/Catalog/
Appendix I. Solubility, Stability and Reactivity data as determined by Absorption Systems
Solubility
Solubility in PBS (at pH = 7.4) for ML169 was 0.08 μM.
Stability
Stability (at room temperature = 23 °C) for ML169 in PBS (no antioxidants or other protectorants and DMSO concentration below 0.1%) is shown in the table below. After 48 hours, the percent of parent compound remaining was not reported, but the assay variability over the course of the experiment ranged from a low of 89% (at 15 minutes) to a high of 142% (at 1 hour).
| Percent Remaining (%) | |||||||
|---|---|---|---|---|---|---|---|
| Compound | 0 Min | 15 Min | 30 Min | 1 Hour | 2 Hour | 24 Hour | 48 Hour |
| ML169 | 100 | 89 | 99 | 142 | 91 | 103 | --- |
Appendix II. Liquid Chromatography-Mass Spectrometry (LCMS) and Nuclear Magnetic Resonance (NMR) as prepared by Vanderbilt Specialized Chemistry Center




Element Composition Report

Footnotes
- *
Solubility (PBS at pH = 7.4), Stability and Reactivity experiments were conducted at Absorption Systems. For additional information see: https://www
.absorption.com/site
- Probe Structure & Characteristics
- Recommendations for the scientific use of this probe
- Assay Implementation and Screening
- Bibliography
- Solubility, Stability and Reactivity data as determined by Absorption Systems
- Liquid Chromatography-Mass Spectrometry (LCMS) and Nuclear Magnetic Resonance (NMR) as prepared by Vanderbilt Specialized Chemistry Center
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- Review Discovery and development of the a highly selective M(1) Positive Allosteric Modulator (PAM).[Probe Reports from the NIH Mol...]Review Discovery and development of the a highly selective M(1) Positive Allosteric Modulator (PAM).Bridges TM, Lewis LM, Dawson ES, Weaver CD, Lindsley CW. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Discovery of a Highly Selective in vitro and in vivo M1 Allosteric Agonist Probe.[Probe Reports from the NIH Mol...]Review Discovery of a Highly Selective in vitro and in vivo M1 Allosteric Agonist Probe.Lindsley C, Lewis M, Weaver D. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Discovery of the first mAChR 5 (M5) selective ligand, an M5 Positive Allosteric Modulator (PAM).[Probe Reports from the NIH Mol...]Review Discovery of the first mAChR 5 (M5) selective ligand, an M5 Positive Allosteric Modulator (PAM).Bridges TM, Lewis LM, Weaver CD, Lindsley CW. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Discovery of a Highly Selective in vitro and in vivo M4 Positive Allosteric Modulator (PAM).[Probe Reports from the NIH Mol...]Review Discovery of a Highly Selective in vitro and in vivo M4 Positive Allosteric Modulator (PAM).Lewis LM, Bridges TM, Niswender CM, Weaver CD, Lindsley CW. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Development of the First Highly Selective mAChR 5 (M(5)) Positive Allosteric Modulator (PAM).[Probe Reports from the NIH Mol...]Review Development of the First Highly Selective mAChR 5 (M(5)) Positive Allosteric Modulator (PAM).Bridges TM, Lewis LM, Weaver CD, Wood MR, Daniels JS, Conn PJ, Lindsley CW. Probe Reports from the NIH Molecular Libraries Program. 2010
- Discovery and development of a second highly selective M1 Positive Allosteric Mo...Discovery and development of a second highly selective M1 Positive Allosteric Modulator (PAM) - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...