

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

NR5A2 (Liver receptor homologue-1; LRH1) belongs to the four-member NR5A, or Ftz-F1, subfamily V of nuclear receptors. Murine LRH1 was originally identified due to its sequence homology to Drosophila Fushi tarazu factor-1. Orthologs were subsequently identified in other species including rat, chicken, horse, zebrafish and human. LRH1, and its closest family member steroidogenic factor-1 (SF-1, NR5A1), bind to identical DNA consensus sequences (response elements; REs) and both are able to bind phospholipids in their ligand binding domains (LBDs). LRH1 was shown to regulate expression of Cyp19 (aromatase), suggesting LRH1 as a target for inverse agonists for the treatment of ER-positive breast cancers. Interestingly, recently it was shown that LRH1 promotes motility and invasiveness in both ER-positive and ER-negative breast cancer cells (MDA-MB-231), with remodeling of the actin cytoskeleton and E-cadherin processing observed with LRH1 over-expression. These findings suggest that inhibition of LRH1 activity should be useful in attenuating both migration and invasion in ER-positive and ER-negative breast cancers. In addition to LRH1’s role in cholesterol metabolism and bile acid homeostasis, the receptor can impact expression of markers of acute phase response (APR) to tissue injury. Thus, we endeavored to identify potent and selective LRH1 inverse agonists which may provide new approaches for the treatment of cancer and to blunt APR. We pursued a Center-based Initiative with transfection studies testing the ability of various chemical scaffolds to inhibit LRH-1-mediated activation of the Cyp19-Aromatase-luciferase and the StAR-luciferase reporter, followed by counterscreening against SF-1 and VP16, as well as studies examining the effect of compounds on LRH-1 modulation of the APR markers haptoglobin and serum amyloid A1 and A4 (SAA1 and SAA4). Combined, the studies led to identification of two novel LRH-1 inverse agonist probe compounds ML179 (PubChem CID 45100448) and ML180 (PubChem CID 3238389) with potent activity in breast cancer cells. In reporter assays ML179 and ML180 had potency IC50 values of 320nM and 3.7 μM and maximum efficacy of 40% and 64% repression, respectively. It is unclear at this stage if maximum repression is more important than potency; thus two probes were declared that differ from each other in terms of potency and efficacy. Further optimization is focused on improving both potency and efficacy. Also, it is likely that the selectivity of these probe compounds versus SF1 is cell context-and promoter-dependent. The mechanism of action of these probes will require much more detailed studies which are currently underway.
Assigned Assay Grant #: U54 MH084512
Screening Center Name & PI: Scripps Research Institute Molecular Screening Center (SRIMSC), Hugh Rosen
Chemistry Center Name & PI: SRIMSC, Hugh Rosen
Assay Submitter & Institution: Patrick Griffin, The Scripps Research Institute (TSRI)
Summary Bioassay Identifier (AID): 488781
Probe Structure & Characteristics

ML 180
CID 3238389
SID 99344023

ML 179
CID 45100448
SID 92092843
| CID/ML# | Target Name | IC50 (nM) [SID, AID] | Anti-target | IC50 (μM) [SID, AID] | Fold Selective | Secondary Assay(s) Name: IC50 (nM) [SID, AID] |
|---|---|---|---|---|---|---|
| CID 3238389/ML180 | LRH1 (NR5A2) | 3700 nM [SID 99344023, AID 488780] Active | SF1 (NR5A1) | > 10 μM [SID 99344023, AID 488780] Inactive | >2.7 | VP16 Fold Change Counterscreen Assay: 1% [SID 99344023, AID 488775] Inactive Mechanism of Action Assays (LRH1 Target Genes) Haptoglobin QPCR Assay: 0.12 fold change (at 10 μM) [SID 99344023, AID 488769] Active SAA1 QPCR Assay: 0.09 fold change (at 10 μM) [SID 99344023, AID 488769] Active SAA4 QPCR Assay: 0.45 fold change (at 10 μM) [SID 99344023, AID 488769] Active MTT Breast Cancer Cytotoxicity Assay: [SID 99344023, AID 504928] Active Star Promoter Counterscreen: 2.05μM IC50 [SID 99344023, AID 504933] Active Nuclear Receptor Profiling: [SID 99344023, AID 504934], Active |
| CID 45100448/ML179 | LRH1 (NR5A2) | 320nM [SID 92092843, AID 488780] Active | SF1 (NR5A1) | > 10μM [SID 92092843, AID 488780] Inactive | >31.25 | VP16 Fold Change Counterscreen Assay: −6% [SID 92092843, AID 488775] Inactive Mechanism of Action Assays (LRH1 Target Genes) Haptoglobin QPCR Assay: 0.14 fold change (at 10 μM) [SID 92092843, AID 488769] Active SAA1 QPCR Assay: 0.07 fold change (at 10 μM) [SID 92092843, AID 488769] Active SAA4 QPCR Assay: 0.45 fold change (at 10 μM) [SID 92092843, AID 488769] Active Star Promoter Counterscreen: 2.12μM IC50 [SID 92092843, AID 504933] Active |
Recommendations for scientific use of the probe
• What limitations in current state of the art is the probe addressing? Currently there are no inverse agonists described for LRH1. Whitby and coworkers have reported the identification of cis-bicyclo[3.3.0]-oct-2-enes as synthetic agonists for both SF-1 and LRH1 with one compound in particular having the ability to induce the doubling of mRNA levels of SHP, a downstream target of both receptors, in HepG2 cells [14]. While these synthetic ligands are intriguing, several problems remain. First, the functional activity of these compounds was determined using a FRET-based biochemical coactivator recruitment assay using small peptides representing the LXXLL motifs of the cofactor; for LRH1 a peptide derived from TIF2 (SRC2) was used and for SF-1 a 23 amino acid peptide fragment of DAX-1 was used. The complimentary cellular activity of these compounds was described for only one compound. Second, the most potent and efficacious agonists lacked functional selectivity over SF-1. The paper describes two partial agonists of LRH1 that are devoid of SF-1 activity showing promise for the ability to obtain functional selectivity. Perhaps more important was the discovery of a potent partial agonist of SF-1 that was devoid of LRH1 activity, strongly suggesting the ability to discover potent and selective agonists within the NR5A subfamily. To test the utility of these compounds, we synthesized two of the most potent compounds described in the Whitby manuscript and evaluated these molecules for their ability to modulate LRH1 in a cell-based assay. Prior to testing, the structures of these compounds were confirmed by NMR and their high purity was confirmed by LC-MS. Unfortunately, these compounds, although potent in a biochemical assay, appear to be cytotoxic even at modest concentrations. Therefore, the discovery of potent, cell active LRH1 modulators remains a high priority in elucidating the tissue-specific functions of LRH1 and its role in mammalian physiology.
Overall the goals of the LRH1 modulator program are to identify potent and selective in vivo active LRH1 agonists and inverse agonists. Obviously, we need to better understand the function of this important receptor and its role in diseases like breast cancer if we are going to development new treatments. To do that, first we need to accelerate the identification of chemical probes. Prior to the work described in this Probe Report, there were no potent in vivo active LRH1 agonists and no reports of any inverse agonists. Thus, probes ML180 and ML179 are first in class LRH1 inverse agonists and these probes constitute significant contributions to the field.
Additional mechanism of action (MOA) studies are required to determine if inverse agonists of LRH1 function by modulating the receptors activity directly by either displacing co-activators or by recruiting co-repressors. It is also possible that these compounds modulate the post-translational status of the receptor directly by impacting recruitment of transcriptional machinery by affecting the receptors’ occupancy at promoters of LRH1 target genes. PTMs could also modulate the receptor such that it trans-represses target genes in a DNA binding independent fashion.
• What will the probe be used for? Probes ML180 and ML179 can be used in cell biological studies to elucidate the role of LRH1 in metabolic diseases, and tumorigenesis. Recently, LRH1 has been shown to play a role in the transcriptional regulation of pathways involved in cancer. Simpson and coworkers were the first to report high levels of LRH1 expression in preadipocytes and they correlated the expression of LRH1 with transcriptional activation of the aromatase cytochrome p450 (CYP19) gene [1]. Aromatase expression has been shown to be highly upregulated in breast tumors as well as breast adipose tissue surrounding tumors. The hallmark of upregulation of CYP19 expression is a switch in promoters from the normal adipose-specific promoter I.4 to the gonadal type PII promoter on the CYP19 gene [2–4]. The Cyp19PII promoter is cAMP-dependent and stimulators such as prostaglandin E2, forskolin, and PMA dramatically induce LRH1-dependent Cyp19PII expression up to, in some reports, 500 fold. This activity was further induced in a synergistic fashion by coexpressing LRH1 and the transcription factors GATA3 and GATA4 in the presence of a CYP19PII luciferase reporter plasmid following stimulators of the cAMP pathway [5]. Moreover, this increase in aromatase expression is completely abrogated following overexpression of SHP which is a negative regulator of LRH1 activity [6, 7]. It has been shown that upregulation of aromatase expression in breast adipose tissue surrounding tumors is responsible for the 10–50 fold increase in estrogen produced in these cells [8]. Unfortunately, this provides fertile ground for the growth and spread of estrogen-dependent tumors which comprise nearly 75% of all breast cancer occurrences in post-menopausal women [1, 9]. Aromatase inhibitors are used clinically in breast cancer treatment but unfortunately these compounds reduce estrogen production in other tissues giving rise to significant side effects such as bone loss. Therefore, inhibitors or inverse agonists of LRH1-dependent activation of aromatase expression could represent a strategy for the development of novel breast-specific tumor therapies.
In addition to modulating expression of the aromatase gene in breast tumors and surrounding tissues, LRH1 has been implicated in cell proliferation and intestinal cancer. Murine hepatic and pancreatic cells overexpressing LRH1 more than doubled in growth rate and formed colonies in soft agar [10]. LRH1 was shown to mediate these effects by acting in synergy with β-catenin to transactivate CyclinD1 and CyclinE1. Furthermore, these effects were reversed by introducing LRH1 siRNA or by overexpression of SHP demonstrating the functionally specific role of LRH1 in cell proliferation. These studies were further extended to examine the effects of LRH1 expression on intestinal tumorigenesis as LRH1 is highly expressed in rapidly dividing intestinal crypt cells. Auwerx and coworkers examined the effects of reduced LRH1 expression on intestinal tumorigenesis using two independent mouse models of the disease [10]. APCmin/− mice that were made haploinsufficient for LRH1 showed lower LRH1 expression and dramatically less tumor formation than mice with wild-type levels of LRH1 expression. In addition, mice lacking one LRH1 allele that were treated with AOM, a chemical inducer of colon cancer, had significantly reduced aberrant crypt foci than mice that expressed normal levels of LRH1.
More recently, overexpression of LRH1 in both ER positive and ER negative cell lines has been shown to promote motility and cell invasiveness in both ER-positive as well as ER-negative breast cancer cells (MDA-MB-231), with remodeling of the actin cytoskeleton and E-cadherin processing observed [11]. This observation suggests that inverse agonists of LRH1 would also be useful in treating ER negative breast cancer. We present data showing the ability of ML180 to inhibit the growth of MDA-MB-231 cells as well as other ER negative and ER positive cancer cell lines.
• Who in the research community will use the probe? Probes ML180 and ML179 can be used by academic researchers studying cell biology, molecular biology, and tumor biology. In addition, the two identified probes will be useful for any researcher working in the field of metabolism, cancer biology, APR and drug discovery.
• What is the relevant biology to which the probe can be applied? Most nuclear receptors function as homodimers or as heterodimers with the retinoid X receptor (RXR) to bind their cognate DNA response elements in promoter regions of target genes [12–14]. However, members of the NR5A subfamily such as SF-1 and LRH1 bind DNA with high affinity as monomers through interactions between the Ftz-F1-consensus binding site on target genes and the Ftz-F1 box, which is a 26 amino acid stretch at the C-terminus of the DNA binding domain (DBD) of these receptors [15]. Additionally, most nuclear receptors require binding of ligand to become transcriptionally active. The model of activation for most nuclear receptors suggests that ligand binding induces conformational changes in the LBD of the receptor that leads to recruitment of coactivators and basal transcriptional machinery with subsequent activation of target genes. Interestingly, members of the NR5A subfamily appear to be constitutively active when expressed in cells. Recently a number of laboratories have identified the presence of phospholipids in the ligand binding pockets of both SF-1 and LRH1 and their presence leads to the recruitment of coactivators in vitro [16–18]. Whether phospholipids are in fact endogenous ligands of NR5A subfamily receptors remains undetermined. The possibility that the NR5A family represent ligand-independent transcription factors does exist. However, a recent publication by Whitby and coworkers [19] revealed small molecule agonists for both LRH1 and SF-1. These compounds were characterized in a biochemical assay utilizing purified protein. The most potent LRH1 agonist described lacked functional selectivity over SF-1 although a potent partial agonist of SF-1 lacking LRH1 activity was discovered. Partial agonists of LRH1 (EC50 1.2 μM, 45% relative efficacy) were also described that lacked affinity for SF-1. More importantly, we synthesized and characterized several examples from the Whitby manuscript and these compounds, although potent in a biochemical assay, were cytotoxic even at modest concentrations (~10 μM).
Perhaps more important is a recent publication resulting from our MLPCN screening center describing a HTS campaign for SF-1 [20]. This screen revealed that chemically tractable small molecule inverse agonists of SF-1 can be discovered directly from an HTS campaign. These compounds were inactive in the RORα counterscreen indicating that they do not inhibit luciferase and they are not potent cytotoxic agents. These studies, combined with the Whitby publication, support the notion that potent and selective small molecule agonist and inverse agonists of LRH1 can be discovered.
Crystal structures for both SF-1 and LRH1 have been solved and these structures have provided some clues to the constitutive activity of these receptors [17, 21, 22]. Most nuclear receptors undergo changes in the conformational dynamics of the ligand binding pocket upon ligand binding resulting in stabilization of the AF2 (activation function 2) surface of the coactivator binding interface of the receptors creating a charge clamp to facilitate recruitment and binding of coactivators [12, 14]. In contrast, the crystal structures of SF-1 and LRH1 show that this stabilization of the coactivator interacting region of the LBD’s may occur as a result of a unique fourth sandwich layer formed by helix 2 (H2) [17, 22]. This region of H2 comes into close proximity with H12 in the absence of ligand to constitutively stabilizing AF2 facilitating coactivator binding. This structural feature of the receptor is also present in the murine receptor structure that was obtained with an empty ligand binding pocket. As mentioned above, the crystal structures of both human and mouse SF-1 as well as human LRH1 revealed the presence of bacterial phospholipids in the ligand binding pockets of these receptors. While the exact role of phospholipids in modulating the activity of these receptors is controversial, these findings further confirm that the NR5A receptors are capable of binding ligands that could potentially modulate their activity in vivo.
Relevance to Human Health: In addition to the role of LRH1 in tumorigenesis discussed above, LRH1 has been shown to play a pivotal role in cholesterol metabolism and bile acid homeostasis in the liver [23]. LRH1 transcriptionally regulates cholesterol 7α-hydroxylase (CYP7A1) [24], the rate-limiting enzyme of the bile acid biosynthetic pathway along with sterol 12 α-hydroxylase (CYP8B1) [25], which is required for cholic acid production. Moreover, LRH1 regulates transcription of a number of key enzymes involved in the uptake of cholesterol from tissues and plasma. Cholesterol ester transfer protein (CETP) is an LRH1 target gene that catalyzes the transfer of cholesterol esters from HDL particles to lipoproteins that can be extracted from serum by the hepatic low density lipoprotein receptor [12, 26]. CETP is currently targeted by several pharmaceutical companies for treatment of hypercholesterolemia, although Pfizer recently withdrew its late stage clinical candidate. It is unclear at this point in time if this is a problem with the specific compound or a problem with the mechanism. Perhaps modulation of the pathway instead of inhibition of one step might provide an approach that is better tolerated. In addition, apolipoprotein A1 is transcriptionally regulated by LRH1 and acts as an acceptor molecule for phospholipids and cholesterol coming from peripheral tissues forming pre-HDL particles. These particles mature and are transferred into hepatocytes by the scavenger receptor class B type I cell surface receptor (SR-BI) which is also transcriptionally regulated by LRH1 [27, 28]. Thus, it is clear that LRH1 modulation could play an important role in cholesterol homeostasis.
LRH1 also activates genes involved in inflammatory responses. Delerive and coworkers, were the first to identify LRH1 as a negative regulator of the hepatic acute phase response [29]. Ectopic expression of LRH1 led to reduced expression of pro-inflammatory genes such as haptoglobin, serum amyloid A and C-reactive protein following stimulation of hepatocytes with proinflammatory cytokines IL-1b and IL-6. In addition, LRH1 activated expression of interleukin-1 receptor antagonist (IL-1RA), a potent anti-inflammatory molecule, following induction by IL-1b and IL-6 as well as intraperitoneal injection of lipopolysaccharide [30]. Moreover, partial deficiencies in LRH1 expression in hepatocytes resulted in decreases in expression of IL-1RA and an exaggerated inflammatory response in vitro and in vivo indicating that LRH1 expression leads to the modulation of anti-inflammatory responses. More recently Venteclef and coworkers reported that the GSK selective synthetic agonist induces sumoylation-dependent recruitment of LRH1 to hepatic acute phase response (APR) promoters and this prevents clearance of the corepressor N-CoR resulting in repression of APR genes [31]. Preliminary studies in our lab using Huh7 cells stimulated with IL1β and IL6 results in significant increase in expression of LRH1, haptoglobin (Hp), serum amyloid A-4 (SAA1), and serum amyloid A-4 (SAA4) as determined by qPCR. Surprisingly and in contrast to the anticipated outcome, treatment of these cells with the inverse agonists ML180 and ML179 represses the expression of three of these genes (Hp, SAA1, SAA4) to the level of control cells (unstimulated) and LRH1 to levels below that in control cells. This finding confirms that our inverse agonists can repress the expression of LRH1 and more importantly, suggests that LRH1 inverse agonists can repress APR genes in similar fashion to LRH1 agonists. Clearly the mechanism of action of inverse agonists in this model is likely different than that of agonists. This finding warrants further mechanistic studies which will be carried out as part of this extended probe development project.
1. Introduction
The goal of this project is to identify modulators (agonists and inverse agonists) of the orphan nuclear receptor LRH1, which has been implicated in cancer by enhancing proliferation and cell cycle progression and metabolic disorders through its regulation of genes involved in cholesterol and bile acid homeostasis. In this specific Probe Report we focus on the discovery and characterization of inverse agonists of LRH1.
NR5A2 or Liver receptor homologue-1 (LRH1) is a member of the NR5A, or Ftz-F1, subfamily V nuclear receptors of which there are four members [12]. Murine LRH1 was originally identified due to its sequence homology to the Drosophila Fushi tarazu factor-1 but orthologs have been subsequently identified in several other species including rat, chicken, horse, zebrafish and human [32–37]. LRH1, and its closest family member steroidogenic factor-1 (SF-1, NR5A1), bind to identical DNA consensus sequences (response elements or REs) and both have the ability to bind phospholipids in their ligand binding domains (LBDs) [16–18]. However, LRH1 and SF-1 are expressed in different tissues and thus are considered likely to have non-overlapping, non-redundant functions. SF-1 expression is confined to steroidogenic tissues and adrenals where it regulates development, differentiation, steroidogenesis and sexual determination [13, 35, 37]. LRH1 is highly expressed in tissues of endodermal origin and its expression is essential for normal liver, intestine, and pancreas function. LRH1 has also been shown to be expressed in the ovary and adipose tissue.
In a very recent report, Chand and colleagues investigated the mechanism of action of LRH1 in invasive breast cancer cells [38]. They found that LRH1 promotes motility and cell invasiveness in both ER-positive (MCF-7) and ER-negative (MDA-MB-231) breast cancer cells and similar effects were observed in non-tumorigenic mammary epithelial cells. Interestingly, both remodeling of the actin cytoskeleton and E-cadherin processing were observed when LRH1 was over-expressed. These findings implicate LRH1 in promotion of migration and invasion in breast cancer independent of estrogen sensitivity. Together these findings provided strong evidence that LRH1 plays a significant role in tumor formation both in vitro and in vivo. Therefore, the identification of potent and selective LRH1 inverse agonists may provide new approaches for the treatment of cancer.
2. Materials and Methods
The SRIMSC Center Driven Research Project (CDRP) titled “Functional genomics approaches to small molecule discovery” was recently renewed. This project is focused on High throughput Functional Genomics and our overall goal is to continue to apply this platform towards pathway discovery and selectivity profiling of chemical probes emerging from the MLPCN network. During Years 1 and 2 of this MLPCN Center Driven Research Project we designed and constructed a nuclear receptor (NR) library using resources within the TSRI high-throughput cDNA screening platform. Using a combination of small molecule libraries and these novel GAL4 tagged libraries, we profiled signaling pathways of several orphan nuclear receptors. During this profiling, we discovered activity on RORA, RORG, and LRH1. These orphan NRs have been implicated in a wide range of diseases and syndromes including metabolic and immune disorders, cancer, and neurological dysfunction. One of these discoveries resulted in submission and approval of a MLPCN probe report (AIDs 1901, 1954, 2117, 2139) describing the first synthetic modulators of RORA and RORG. We demonstrated that these compounds can repress glucose production in hepatocytes, and expression of IL17 from TH17 cells, which clearly demonstrated the utility of these chemical probes in models of diabetes and autoimmune disease. A second project within the CDRP was to characterize synthetic inverse agonists of LRH1. The significance of these compounds is described in detail above. As a result, several assays were performed in order to identify novel inverse agonists of LRH1. The specific assays are listed in Table 1.
Table 1
Summary of Performed Assays.
2.1. Assays
The specific descriptions and protocols for each assay can be found in this section and in PubChem.
AID 485348 and 488782: LRH1 Inhibition Assays (single point and dose response)
Name
Center Based Initiative to identify novel inverse agonists of the liver receptor homolog-1 (LRH1; NR5A2): Luminescence-based single point and dose response assays to identify LRH1 inhibitors (Cyp19 aromatase-luciferase reporter 3X%INH and 3XIC50).
Assay Overview
The purpose of these assays is to determine whether powder samples of possible LRH1 inverse agonist probe candidates can inhibit the activity of LRH1. In these assays, HEK293T cells, co-transfected with a full length LRH1 construct in a pSport6 vector backbone (pS6-LRH1) and a Cyp19-Aromatase-luciferase reporter construct, are incubated for 20 hours with test compound. Tissue-specific expression of Cyp19 is regulated by hormones (specifically estrogens) and is increased in response to breast tumor derived factors. LRH1 has been shown to bind to the Cyp19 promoter and regulates its expression in adipose tissue. As designed, a compound that inhibits LRH1 activity will prevent activation of the pS6-LRH1 construct, thereby preventing LRH1-mediated activation of the Cyp19-Aromatase-luciferase reporter, leading to a decrease in well luminescence. Compounds were tested in triplicate at a nominal concentration of 10 micromolar, and in triplicate using a 9-point dose response series starting at a nominal concentration of 10 micromolar.
Protocol Summary
Luciferase reporter assays were conducted using a pSport6 full-length LRH1 construct and Cyp19 aromatase luciferase reporter cotransfected into HEK293T cells. Reverse transfections were performed in bulk using 3×106 cells in 10 cm plates,7μg of total DNA and FuGene6 (Roche) in a 1:3 DNA: lipid ratio. Following 24 hour bulk transfection, cells from were counted and re-plated in 384-well plates at a density of 10,000 cells/well. Following 4 hour incubation, cells were treated with DMSO/compounds for 20 hours. The luciferase levels were measured by addition of BriteLite Plus (Perkin Elmer). Data was normalized to luciferase signal from DMSO treated cells. The fold-change inhibition for each compound was calculated as follows:
Cells_treated_with_Test_Compound / Cells_treated_with_Vehicle (DMSO).
The average fold-change of each compound tested was calculated.
PubChem Activity Outcome and Score
Any compound that exhibited a fold-change inhibition greater than the hit cutoff calculated (> 25%Inhibition) was declared active.
For dose response assays, each test compound’s percent inhibition was plotted against compound concentration. A four-parameter equation describing a sigmoidal dose-response curve was then fitted with adjustable baseline using GraphPad Prism software. The reported IC50 values were generated from fitted curves by solving for the X-intercept value at the 50% inhibition level of the Y-intercept value. In cases where the highest concentration tested (i.e. 10 micromolar) did not result in greater than 50% inhibition, the IC50 value was determined manually as greater than 10 micromolar. Compounds with an IC50 value greater than 5 micromolar were considered inactive. Compounds with an IC50 value equal to or less than 5 micromolar were considered active. Activity score was ranked by the potency of the compounds, with the most potent compounds assigned the highest activity scores.
List of Reagents
LRH1 and Cyp19 plasmid DNAs (assay provider lab)
384-well plates (PerkinElmer, part 6007688)
Britelite Plus (PerkinElmer, part 6016767)
DMEM (Mediatech Inc, Part 10 013 CV)
Fugene 6 (Roche Applied Science, part 11814443001).
AID 488779 and 488780: SF1 Inhibition Counterscreens (single point and dose response)
Name
Center Based Initiative to identify novel inverse agonists of the liver receptor homolog-1 (LRH1; NR5A2): Luminescence-based dose response and single point counterscreen assays to identify inhibitors of the steroidogenic factor-1 (SF1) (3X%INH and 3XIC50).
Assay Overview
The purpose of this assay is to determine whether powder samples of possible LRH1 inverse agonist probe candidates are nonselective due to inhibition of another nuclear receptor, SF-1. This assay also serves to determine SF-1 inhibitory dose response curves for compounds In this assay, HEK293T cells, co-transfected with a full length SF-1 construct in a pSport6 vector backbone (pS6-SF-1) and a 5xSFRE-luciferase reporter construct, are incubated for 20 hours with test compound. As designed, a compound that inhibits SF-1 activity will prevent activation of the pS6 SF-1 construct, thereby preventing SF-1-mediated activation of the 5xSFRE-luciferase reporter, leading to a decrease in well luminescence. Compounds were tested in triplicate using a 9-point dose response series starting at a nominal concentration of 10 micromolar.
Protocol Summary
Luciferase reporter assays were conducted using a pSport6 full-length SF-1 construct and 5xSFRE luciferase reporter cotransfected into HEK293T cells. Reverse transfections were performed in bulk using 3×106 cells in 10 cm plates,7μg of total DNA and FuGene6 (Roche) in a 1:3 DNA: lipid ratio. Following 24 hour bulk transfection, cells from were counted and re-plated in 384-well plates at a density of 10,000 cells/well. Following 4 hour incubation, cells were treated with DMSO/compounds for 20 hours. The luciferase levels were measured by addition of BriteLite Plus (Perkin Elmer). Data was normalized to luciferase signal from DMSO treated cells. The percent inhibition for each compound was calculated as follows:
Cells_treated_with_Test_Compound / Cells_treated_with_Vehicle (DMSO)
PubChem Activity Outcome and Score
Any compound that exhibited a fold-change inhibition less than the hit cutoff calculated (> 25 %Inhibition) was declared active. For dose response assays, each test compound, percent inhibition was plotted against compound concentration. A four-parameter equation describing a sigmoidal dose-response curve was then fitted with adjustable baseline using GraphPad Prism software. The reported IC50 values were generated from fitted curves by solving for the X-intercept value at the 50% inhibition level of the Y-intercept value. In cases where the highest concentration tested (i.e. 10 micromolar) did not result in greater than 50% inhibition, the IC50 value was determined manually as greater than 10 micromolar. Compounds with an IC50 value greater than 5 micromolar were considered inactive. Compounds with an IC50 value equal to or less than 5 micromolar were considered active.
List of Reagents
SF-1 and SFRE plasmid DNAs (assay provider lab)
384-well plates (PerkinElmer, part 6007688)
Britelite Plus (PerkinElmer, part 6016767)
DMEM (Mediatech Inc, Part 10 013 CV)
Fugene 6 (Roche Applied Science, part 11814443001).
AID 488775: VP16 Inhibition Counterscreen (Single Point Fold Change)
Name
Center Based Initiative to identify novel inverse agonists of the liver receptor homolog-1 (LRH1; NR5A2): Luminescence-based counterscreen assay to identify inhibitors of the human herpes virus VP16 transcriptional activator protein (VP16) (3X%INH).
Assay Overview
The purpose of this assay is to determine whether powder samples of possible LRH1 inverse agonist probe candidates are nonselective due to inhibition of VP16. In this counterscreen assay the nuclear receptor plasmid was replaced by the GAL4DBD-VP16LBD plasmid, which expresses the strong transactivation domain of the herpes simplex virus Virion Protein 16 (VP16) fused to the GAL4 DBD. Cells are co-transfected with the 5xGAL4 response element (UAS) luciferase reporter to monitor GAL4DBD-VP16LBD activity, followed by incubation with test compounds for 18–24 hours. As designed, compounds that inhibit VP16 activity will decrease pGAL4DBD-VP16LBD activity, leading to reduced activation of the pG5-luc and decreased well luminescence. These compounds are likely to be nonselective inhibitors or cytotoxic. Compounds were tested in triplicate at a nominal concentration of 5 micromolar.
Protocol Summary
Luciferase reporter assays were conducted using a pBind GAL4DBD-VP16LBD construct and UAS luciferase reporter cotransfected into HEK293T cells. Reverse transfections were performed in bulk using 4×106 cells in 10 cm plates, 9μg of total DNA and FuGene6 (Roche) in a 1:3 DNA: lipid ratio. Following 24 hour bulk transfection, cells from were counted and replated in 384-well plates at a density of 10,000 cells/well. Following 4 hour incubation, cells were treated with DMSO/compounds for 20 hours. The luciferase levels were measured by addition of BriteLite Plus (Perkin Elmer). Data was normalized to luciferase signal from DMSO treated cells. The fold-change inhibition for each compound was calculated as follows:
Cells_treated_with_Test_Compound / Cells_treated_with_Vehicle (DMSO).
PubChem Activity Outcome and Score
Any compound that exhibited a fold-change inhibition less than the hit cutoff calculated (> 10 %Inhibition) was declared active.
List of Reagents
VP16 and GAL4 UAS plasmid DNAs (assay provider lab)
384-well plates (PerkinElmer, part 6007688)
Britelite Plus (PerkinElmer, part 6016767)
DMEM (Mediatech Inc, Part 10 013 CV)
Fugene 6 (Roche Applied Science, part 11814443001).
AID 488769: QPCR mRNA Fold Change Assay (Haptoglobin, SAA1, SAA4 gene targets)
Name
Center Based Initiative to identify novel inverse agonists of the liver receptor homolog-1 (LRH1; NR5A2): fluorescence-based cell-based quantitative PCR assay to identify inhibitors of LRH1 target gene expression.
Assay Overview
The purpose of this assay is to determine whether powder samples of possible LRH1 inverse agonist probe candidates are able to block the expression of pro-inflammatory target genes haptoglobin, SAA1, and SAA4. LRH1 activation has been shown to prevent the cytokine induced stimulation of certain proinflammatory genes from the liver that are activated in the acute phase response. The genes are Haptoglobin, SAA1 and SAA4. In these assays huh7 cells endogenously expressing LRH1 were treated with 10micromolar compound or DMSO vehicle for 18 hours. Following compound incubation, 3nM IL1β and IL6 inflammatory cytokines were added to cells and incubated for a further 3 hours followed by isolation of RNA, conversion to cDNA, and Taqman-based QPCR. As designed, a compound that inhibits LRH1 activity will reduce target gene expression following addition of cytokines, leading to decreased production of the PCR amplicon, thereby reducing fluorescence, and increasing Ct.
Protocol Summary
Huh7 cells endogenously expressing LRH1 were plated in 6 well plates at a density of 200,000 cells/well and after 18 hour incubation, treated with 10micromolar compound or DMSO vehicle for additional 18 hours. Following compound incubation, 3nM IL1b and IL6 inflammatory cytokines were added to cells and incubated for a further 3 hours. Then cells were lysed and RNA was isolated using the RNeasy kit (QIAGEN, Valencia, CA). DNA was generated using the Taqman reverse transcription kit (Applied Biosystems, Foster City, CA). Quantitative real-time polymerase chain reaction (PCR) was performed in triplicate using LightCycler RNA Amplification Kit HybProbe master mix (Roche) with Taqman MGB Probe 6FAM--MGBNFQ on a model LightCycler480 real time PCR system (Roche). Data are expressed as the mean percent inhibition plus or minus SD of 3 replicates normalized to 100μg total RNA. The percent inhibition was normalized based on measurement of total RNA.
PubChem Activity Outcome and Score
Any compound that exhibited a fold-change inhibition less than the hit cutoff calculated (> 25 %Inhibition) was declared active.
List of Reagents
384-well plates (PerkinElmer, part 6007688)
Britelite Plus (PerkinElmer, part 6016767)
DMEM (Mediatech Inc, Part 10 013 CV)
Fugene 6 (Roche Applied Science, part 11814443001).
QiaShredder (Qiagen, 79656)
RNeasy mini kit (Qiagen, 74104)
LightCycler RNA Amplification Kit HybProbe (Roche, 12015145001)
Forward primer (Lifetech Applied Biosystems)
Reverse primer (Lifetech Applied Biosystems)
MGBProbe (Lifetech Applied Biosystems, 4304971, 6FAM)
LightCycler480 multiwell plate 96 (Roche, 04729692001)
LightCycler480 sealing foil (Roche, 04729757001)
Specific Primer Sequences are as follows:
Haptoglobin-For-5′-AATGTGAAGCAGATGACG-3′
Haptoglobin-Rev-5′-GGGCAATGTCTTTCGCTGT-3′
SAA1-For-5′-CTGCAGAAGTGATCAGCG-3′
SAA1-Rev-5′-ATTGTGTACCCTCTCCCC-3′
SAA4-For-5′-CCAGTGAAAGCTGGCGTT-3′
SAA4-Rev-5′-GAGAAGTGTGTGGCTCACAGCC-3′
AID 504933: Star Inhibition Counterscreen (dose response)
Name
Late stage assay provider results from the probe development effort to identify inverse agonists of the liver receptor homolog-1 (LRH-1; NR5A2): luminescence-based cell-based assay to identify inhibitors of the StAR (Steroidogenic acute regulatory protein).
Assay Overview
The purpose of this assay is to determine whether powder samples of possible LRH1 inverse agonist probe candidates can inhibit the activity of LRH1, as measure by inhibition of promoter activity of the Steroidogenic acute regulatory protein (Star). In this assay, HEK293T cells, co-transfected with a full length LRH1 construct in a pSport6 vector backbone (pS6-LRH1) and a Star-luciferase reporter construct are incubated for 20 hours with test compound. StAR is a transport protein that regulates cholesterol transfer within the mitochondria, which is the rate-limiting step in the production of steroid hormones. It is primarily present in steroid-producing cells, including theca cells and luteal cells in the ovary, Leydig cells in the testis and cell types in the adrenal cortex. Compounds were tested in triplicate at a nominal concentration of 10 micromolar, and in triplicate using a 9-point dose response series starting at a nominal concentration of 10 micromolar.
Protocol Summary
Luciferase reporter assays were conducted using a pSport6 full-length LRH1 construct and StAR luciferase reporter cotransfected into HEK293T cells. Reverse transfections were performed in bulk using 3×106 cells in 10 cm plates, 9μg of total DNA and X-tremeGENE 9 DNA Transfection Reagent in a 1:3 DNA: lipid ratio. Following 24 hour bulk transfection, cells from were counted and re-plated in 384-well plates at a density of 8,000 cells/well. Following 4 hour incubation, cells were treated with DMSO/compounds for 20 hours. The luciferase levels were measured by addition of BriteLite Plus Reagent. Data was normalized to luciferase signal from DMSO treated cells. The fold-change inhibition for each compound was calculated as follows:
Cells_treated_with_Test_Compound / Cells_treated_with_Vehicle (DMSO).
Then the average percent maximal response and IC50 of each compound tested were calculated.
PubChem Activity Outcome and Score
Any compound that exhibited a fold-change inhibition greater than the hit cutoff calculated (> 25%Inhibition) was declared active. For dose response assays, each test compound, percent inhibition was plotted against compound concentration. A four parameter equation describing a sigmoidal dose-response curve was then fitted with adjustable baseline using GraphPad Prism software. The reported IC50 values were generated from fitted curves by solving for the X-intercept value at the 50% inhibition level of the Y-intercept value. In cases where the highest concentration tested (i.e. 10 micromolar) did not result in greater than 50% inhibition, the IC50 value was determined manually as greater than 10 micromolar. Compounds with an IC50 value greater than 5 micromolar were considered inactive. Compounds with an IC50 value equal to or less than 5 micromolar were considered active. For PubChem, activity score was ranked by the potency of the compounds, with the most potent compounds assigned the highest activity scores.
List of Reagents
LRH1 and StAR plasmid DNAs (assay provider lab)
384-well plates (PerkinElmer, part 6007688)
Britelite Plus (PerkinElmer, part 6016767)
DMEM (Mediatech Inc, Part 10 013 CV)
X-tremeGENE 9 DNA Transfection Reagent (Roche Applied Science, part 06365809001).
AID 504934: Nuclear Receptor Profiling Counterscreen
Name
Late stage assay provider results from the probe development effort to identify inverse agonists of the liver receptor homolog-1 (LRH-1; NR5A2): luminescence-based high throughput cell-based assay to identify modulators of human nuclear receptors.
Assay Overview
The purpose of this assay is to identify compounds that act as modulators of human nuclear receptors and to demonstrate the utility of the GAL4 nuclear receptor library (13). This assay screens endogenous and synthetic ligands against a GAL4 nuclear receptor library which was built by replacing the endogenous N-terminus and DNA-binding domain (DBD) of all 48 receptors with a GAL4 DBD. The fusion constructs consist of the GAL4 DBD, the hinge domain, ligand binding domain (LBD), and F domain if applicable, of the human receptors. Plasmids coding for full-length receptors were also included for some receptors. In this assay HEK293T cells are co-transfected with a single GAL4 receptor and a luciferase reporter containing an upstream activating sequence (UAS) recognized by the GAL4 DBD, followed by treatment with test compounds. As designed, compounds that modulate activity of a particular nuclear receptor will modulate the binding of the GAL4 DBD to the UAS, thereby modulating luciferase production, resulting in an increase or decrease in well luminescence. Compounds are tested in triplicate at a nominal test concentration of 2 micromolar.
Protocol Summary
The nuclear receptor library was plated into 384-well plates. HEK293T cells were reverse transfected with the well-specific construct and the UAS luciferase reporter pGL4.31 using Fugene6 transfection reagent in a final volume of 40 microliters. Control wells containing constructs encoding for the GAL4 DBD alone (pBind) or GAL4 fused to VP16 were also analyzed. After 24 hours, optimized compounds (2 μM final concentration) or DMSO was added to the plates and allowed to incubate for 20 hours. Next, 40 microliters of BriteLite was added to all wells and luciferase activity was measured on the PerkinElmer Envision 2104. Compounds that attenuate the GAL4-VP16-dependent luciferase activity in the positive control are considered promiscuous or cytotoxic. Each compound was evaluated using two plates of the GAL4 NR library providing six replicates and was normalized to DMSO and VP16 well controls. Compounds with mean signals three standard deviations from the DMSO controls were considered hits in this assay.
List of Reagents
Fugene6 transfection reagent (Roche Applied Sciences)
BriteLite reagent (Perkin Elmer)
pGL4.31 construct (Promega)
384-well plates (Greiner, part 789176)
AID 504928: Breast Cancer Cell Cytotoxicity Counterscreen (MTT)
Name
Late stage assay provider results from the probe development effort to identify inverse agonists of the liver receptor homolog-1 (LRH-1; NR5A2): absorbance-based cell-based assay to identify cytotoxic compounds in various cell types.
Assay Overview
The purpose of this assay is to determine cytotoxicity dose response curves for powder samples of possible LRH1 inverse agonist probes candidates, using a variety of tumor cell lines. This assay is based upon the reduction of the yellow tetrazolium salt (MTT) in metabolically active cells to form insoluble purple-blue formazan crystals, which are solubilized by the addition of a detergent. MTT reduction occurs inside cells via the action of mitochondrial dehydrogenases. Formazan production is directly proportional to cell number, and metabolically inactive cells produce low levels of formazan. All values were normalized to DMSO negative control. As designed, a compound that inhibits cell growth or proliferation or is directly cytotoxic, will lead to decreased production of purple formazan crystals, thereby reducing well absorbance. Some compounds were tested in duplicate using a 5-point dilution series starting at a nominal concentration of 10 micromolar. For some assays, doses tested were 10.0, 3.3, 1.1, 0.37, 0.123, 0.041, 0.0137, and 0.0045 micromolar. Control treatment of cells was media alone.
Protocol Summary
A selection of some of the possible cell types tested in this assays are listed here: Human B lymphocyte (Lymphoma) Raji cells, Mouse B lymphocyte EuMyc Lymphoma cells, Mouse Pre-B lymphocyte (cancer) 70Z/3 cells, 9×1010 cells, Mouse T lymphocyte (Leukemia) Jurkat cells, Human Breast Cancer MCF7 cells, Human Breast Cancer T47D cells, Human Breast Cancer MD-MDA-231 cells, Human breast (non-cancer) HS578 cells, Human Breast Cancer SKBR cells.
Cells were seeded in 96-well plate based on the standard cell concentrations, 50 μL per well. Cells were cultured in 37°C, 5%CO2 for 3–5 days. Add MTT (Chemicon International) 10 μL per well, continue culturing for 4 hours. Add isopropanol/0.04N HCL, 100 μL per well, pipit up and down to dissolve the formazan into a homogeneous blue solution. Read at 570nm and 630nm. Use dual wavelength of 570nm subtracted from reference 630 nm. The percent maximal response (inhibition) for each replicate well of each compound was calculated as follows:
[Cells_treated_with_Test_Compound] / [Cells_treated_with_Vehicle (DMSO)]
Then the average percent maximal response and standard deviation of each compound tested were calculated. For selected test compounds, fold inhibition was plotted against compound concentration. Either the reported IC50 or fold change values were calculated from GraphPad Prism software. Compounds with an IC50 greater than 10 μM or a fold change value greater than 0.6 were considered inactive. Compounds with an IC50 equal to or less than 10 μM, or a fold change value less than 0.6 were considered active.
List of Reagents: MTT (purchased from Chemicon International)
2.2. Probe Chemical Characterization
| Compound | SR Number | MLS | CID | SID | Solubility in PBS (μM) | MichaelAcceptor 100 μM GSH trap | Stability in PBS (t1/2 (hr) |
|---|---|---|---|---|---|---|---|
| PROBE #1 (ML180) | SR-01000621848-2 | MLS003153119 | CID 3238389 | SID 99344023 | 8.5 | No | > 48 hr |
| PROBE #2 (ML179) | SR-03000001309-1 | MLS003153122 | CID 45100448 | SID 92092843 | 3.9 | No | > 48 hr |
Graphical representations of the stability of the new probe compounds are shown in Figure 1. The new probes have been tested in numerous cell-based assays, demonstrating their cellular permeability, solubility, and stability.

Figure 1
Probe Stability.
LC-MS/MS
All analytical methods were in MRM mode where the parent ion was selected in Q1 of the mass spectrometer. The parent ion was fragmented and a characteristic fragment ion monitored in Q3. MRM mass spectroscopy methods are particularly sensitive because additional time is spent monitoring the desired ions and not sweeping a large mass range. Methods were rapidly set up using Automaton® (Applied Biosystems), where the compounds were listed with their name and mass in an Excel datasheet. Compounds were submitted in a 96-well plate to the HPLC autosampler and slowly injected without a column present. A narrow range centered on the indicated mass was scanned to detect the parent ion. The software then evaluated a few pre-selected parameters to determine conditions that maximized the signal for the parent ion. The molecule was then fragmented in the collision cell of the mass spectrometer and fragments with m/z larger than 70 but smaller than the parent mass were determined. Three separate collision energies were evaluated to fragment the parent ion and the largest three ions were selected. Each of these three fragment ions was further optimized and the best fragment was chosen. The software then inserted the optimized masses and parameters into a template method and saved it with a unique name that indicated the individual compound being optimized. Spectra for the parent ion and the fragmentation pattern were saved and reviewable later.
Solubility
The solubility of compounds was tested in phosphate buffered saline, pH 7.4. Compounds were inverted for 24 hours in test tubes containing 1–2 mg of compound with 1 mL of PBS. The samples were centrifuged and analyzed by HPLC (Agilent 1100 with diode-array detector). Peak area was compared to a standard of known concentration. In cases when the concentration was too low for UV analysis or when the compound did not possess a good chromophore, LC-MS/MS analysis was used.
Stability
Demonstration of stability in PBS was conducted under conditions likely to be experienced in a laboratory setting. The compound was dissolved in 1 mL of PBS at a concentration of 10 μM, unless its maximum solubility was insufficient to achieve this concentration. Low solubility compounds were tested between ten and fifty percent of their solubility limit. The solution was immediately aliquoted into seven standard polypropylene microcentrifuge tubes which were stored at ambient temperature in a block microcentrifuge tube holder. Individual tubes were frozen at −80°C at 0, 1, 2, 4, 8, 24, and 48 hours. The frozen samples were thawed in a room temperature and an equal volume of acetonitrile was added prior to determination of concentration by LC-MS/MS.
Determination of glutathione reactivity
One μL of a 10 mM compound stock solution was added to 1 mL of a freshly prepared solution of 100 μM reduced glutathione. Final compound concentration was 10 μM unless solubility limited. The solution was allowed to incubate at 37°C for two hours prior to being directly analyzed for glutathione adduct formation. LC-MS/MS analysis of GSH adducts was performed on an API 4000 Q-TrapTM mass spectrometer equipped with a Turboionspray source (Applied Biosystems, Foster City, CA). Two methodologies were utilized—a negative precursor ion (PI) scan of m/z 272, corresponding to GSH fragmenting at the thioether bond, and a neutral loss scan of-129 AMU to detect GSH adducts. This triggered positive ion enhanced resolution and enhanced product ion scans [39, 40].
2.3. Probe Preparation: Synthesis of LRH1 Inverse Agonist Probes and Analogs
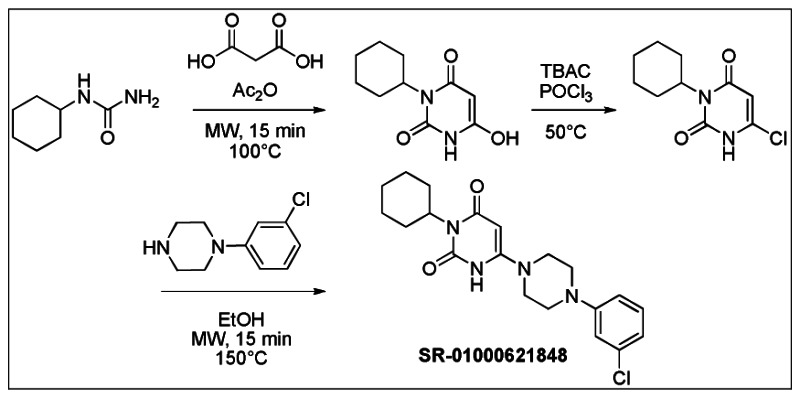
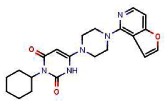
Figure 2Synthesis of SR-01000621848 (Probe 1: ML180) (CID 3238389)
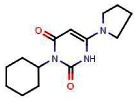
The first reaction was performed using a known method [41]. Cyclohexylurea (510 mg, 3.52 mmol) and malonic acid (370 mg, 3.52 mmol) were weighted into a sealed tube. Acetic anhydride (671 μL, 7.03 mmol) was added and the vial was heated 7 min in a microwave at 100°C. The mixture was then evaporated, EtOH was added and the product precipitated after triturating crude product. The solid was filtrated to provide 300 mg of 3-cyclohexyl-6-hydroxypyrimidine-2,4(1H,3H)-dione (41%) as white powder. 1H NMR (400 MHz, (CD3)2SO): 1.09 (qt, J = 12.9, 3.4 Hz, 1H), 1.25 (qt, J = 12.9, 3.4 Hz, 2H), 1.52–1.64 (m, 3H), 1.76 (broad d, J = 13.0 Hz, 2H), 2.14 (qd, J = 12.5, 3.4 Hz, 2H), 3.58 (s, 2H), 4.41 (tt, J = 12.2, 3.5 Hz, 1H), 11.19 (s, 1H).
The chlorination reaction was performed using a known method [42]. 3-Cyclohexyl-6-hydroxypyrimidine-2,4(1H,3H)-dione (272 mg, 1.29 mmol) and tetrabutylammonium chloride (771 mg, 2.72 mmol) were weighed in a round bottom flask. Phosphorus oxychloride (2.55 mL) was added and the mixture was heated 4h at 50°C, then cooled to ambient temperature and poured in cold water. The aqueous phase was extracted three times by CHCl3. The combined organic extracts were dried over Na2SO4, filtrated and evaporated. The crude residue was purified by silica gel column and eluted with hexane-EtOAc (60/40) to obtain 216 mg of 6-chloro-3-cyclohexylpyrimidine-2,4(1H,3H)-dione (73%) as a white powder. 1H NMR (400 MHz, (CD3)2SO): 1.10 (qt, J = 12.9, 3.2 Hz, 1H), 1.26 (qt, J = 12.9, 3.1 Hz, 2H), 1.47–1.65 (m, 3H), 1.76 (broad d, J = 12.9 Hz, 2H), 2.25 (qd, J = 12.4, 3.3 Hz, 2H), 4.41 (tt, J = 12.1, 3.8 Hz, 1H), 5.82 (s, 1H), 12.21 (s, 1H).
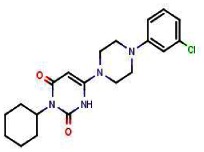
6-Chloro-3-cyclohexylpyrimidine-2,4(1H,3H)-dione (50 mg, 0.22 mmol) and 1-(3-chlorophenyl)piperazine (129 mg, 0.66 mmol) were weighed into a sealed tube. Ethanol (300 μL) was added and the vial was heated 10 min in a microwave at 150°C. The mixture was directly applied to a silica gel column and eluted with CH2Cl2-MeOH (95/5) to obtain 51 mg of SR-01000621848 (61%, purity 99%) as a reddish powder.
FTIR: 2937, 2828, 1691, 1625, 1592, 1430, 1377, 1234, 1195, 1019, 946, 784, 768, 687, 679 cm−1.
1H NMR (400 MHz, (CDCl3)): 1.17 (qt, J = 13.2, 3.4 Hz, 1H), 1.40 (qt, J = 13.2, 3.2 Hz, 2H), 1.62–1.76 (m, 3H), 1.85 (broad d, J = 13.0 Hz, 2H), 2.31 (qd, J = 12.6, 3.1 Hz, 2H), 3.27–3.34 (m, 4H), 3.45–3.53 (m, 4H), 4.78 (bt, J = 12.0 Hz, 1H), 4.99 (bs, 1H), 6.80 (dd, J = 8.3, 2.1 Hz, 1H ), 6.87–6.91(m, 2H), 7.20 (d, J = 8.4 Hz, 1H), 10.33 (s, 1H)
13C NMR (100 MHz, (CDCl3)): 25.6, 26.4, 29.0, 46.0, 48.5, 80.2, 114.4, 116.2, 120.6, 130.3, 135.2, 151.5, 153.3, 164.0.
MS (ES-) m/z = 387 (found for C20H25ClN4O2-H+).

SR-03000001309 (95% last step, purity >98%) obtained as a white powder, was synthesized following the same procedures described for SR-01000621848, replacing in the last step 1-(3-chlorophenyl)piperazine by 1-(3-(trifluoromethyl)phenyl)piperazine.
FTIR: 2935, 2854, 1694, 1626, 1450, 1434, 1389, 1353, 1310, 1232, 1161, 1120, 110, 947, 781, 694 cm−1.
1H NMR (400 MHz, (CDCl3)): 1.16 (qt, J = 13.1, 3.3 Hz, 1H), 1.40 (qt, J = 13.1, 3.3 Hz, 2H), 1.60–1.76 (m, 3H), 1.83 (broad d, J = 13.6 Hz, 2H), 2.31 (qd, J = 12.4, 3.3 Hz, 2H), 3.32–3.39 (m, 4H), 3.49–3.56 (m, 4H), 4.79 (bt, J = 12.1 Hz, 1H), 5.01 (d, J = 2.0 Hz, 1H), 7.08 (dd, J = 8.1, 2.3 Hz, 1H ), 7.10–7.14 (m, 1H), 7.16 (d, J = 7.6 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 10.50 (s, 1H)
13C NMR (100 MHz, (CDCl3)): 25.5, 26.3, 28.9, 46.1, 48.4, 80.2, 112.5 (q, J = 4.4 Hz), 117.1(q, J = 4.1 Hz), 119.2, 124.1 (q, J = 272.7 Hz), 129.8, 131.7 (q, J = 32.1 Hz), 150.6, 153.4, 164.0.
MS (ES-) m/z = 421 (found for C21H25F3N4O2-H+).
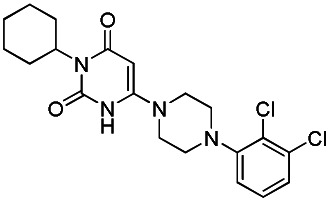
Data for SR-1310 (Probe Analog)
SR-03000001310
SR-03000001310 (97% last step, purity >98%) obtained as a white powder, was synthesized following the same procedures described for SR-01000621848, replacing in the last step 1-(3-chlorophenyl)piperazine by 1-(3-(trifluoromethyl)phenyl)piperazine.
FTIR: 2937, 2856, 1691, 1622, 1575, 1451, 1432, 1423, 1388, 1373, 1241, 1198, 1019, 955, 787 cm−1.
1H NMR (400 MHz, (CDCl3)): 1.12 (qt, J = 13.1, 3.1 Hz, 1H), 1.36 (qt, J = 13.1, 3.1 Hz, 2H), 1.61–1.71 (m, 3H), 1.80 (broad d, J = 13.4 Hz, 2H), 2.30 (qd, J = 12.1, 3.2 Hz, 2H), 3.11–3.20 (m, 4H), 3.48–3.59 (m, 4H), 4.77 (bt, J = 12.2 Hz, 1H), 5.01 (bs, 1H), 6.93 (dd, J = 7.9, 1.6 Hz, 1H ), 7.17 (t, J = 8.0 Hz, 1H), 7.23 (dd, J = 8.1, 1.6 Hz, 1H), 10.29 (s, 1H)
13C NMR (100 MHz, (CDCl3)): 25.5, 26.3, 28.9, 46.5, 50.7, 79.9, 118.5, 125.5, 127.6, 127.7, 134.4, 150.2, 153.2, 153.3, 164.1.
MS (ES-) m/z = 421 (found for C20H24Cl2N4O2-H+).

Data for SR-1174 (Probe Analog)
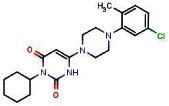
SR-03000001174
SR-03000001174 (97% last step, purity >98%) obtained as a white powder, was synthesized following the same procedures described for SR-01000621848, replacing at the last step 1-(3-chlorophenyl)piperazine by 7-chloro-4-piperazin-1-ylquinoline.
FTIR: 2922, 2848, 1694, 1620, 1575, 1425, 1377, 1232, 1197, 1012, 874, 867, 829, 822, 790, 729 cm−1.
1H NMR (400 MHz, (CDCl3)): 1.05 (qt, J = 13.1, 3.6 Hz, 1H), 1.25–1.42 (m, 2H), 1.57–1.70 (m, 3H), 1.76 (broad d, J = 13.3 Hz, 2H), 2.14 (qd, J = 12.4, 3.1 Hz, 2H), 3.31–3.37 (m, 4H), 3.60–3.67 (m, 4H), 4.77 (bt, J = 11.9 Hz, 1H), 5.04 (d, J = 1.8 Hz, 1H), 6.86 (d, J = 4.9 Hz, 1H ), 7.47 (dd, J = 9.0, 2.2 Hz, 1H), 7.94 (d, J = 9.0 Hz, 1H), 8.09 (d, J = 2.1 Hz, 1H), 8.77 (d, J = 4.9 Hz, 1H), 10.51 (s, 1H)
13C NMR (100 MHz, (CDCl3)): 25.6, 26.3, 28.9, 46.3, 51.6, 53.4, 80.5, 109.1, 121.6, 124.5, 126.8, 129.1, 135.4, 150.1, 151.8, 153.3, 156.0, 163.9.
MS (ES-) m/z = 438 (found for C23H26ClN5O2-H+).

Data for SR-1409 (Probe Analog)
SR-03000001409
SR-03000001409 (97% last step, purity >98%) obtained as a white powder, was synthesized following the same procedures described for SR-01000621848, replacing at the first stage cyclohexylurea by phenylurea and at the last step 1-(3-chlorophenyl)piperazine by 1-(3-(trifluoromethyl)phenyl)piperazine.
FTIR: 3049, 2844, 1726, 1700, 1606, 1495, 1449, 1314, 1232, 1165, 1112, 1099, 1074, 950, 778, 697, 689 cm−1.
1H NMR (400 MHz, (CDCl3)): 2.99–3.04 (m, 4H), 3.36–3.43 (m, 4H), 5.08 (s, 1H), 6.95–7.01 (m, 2H), 7.18 (d, J = 7.8 Hz, 1H), 7.23–7.27 (m, 2H), 7.38–7.44 (m, 2H), 7.44–7.50 (m, 2H), 10.95 (s, 1H).
13C NMR (100 MHz, (CDCl3)): 46.0, 48.0, 79.4, 112.5 (q, J = 4 Hz), 116.9 (q, J = 4 Hz), 119.2, 124.2 (q, J = 272.0 Hz), 128.5, 128.6, 129.1, 129.7, 131.6 (q, J = 31.7 Hz), 134.8, 150.6, 153.5, 163.5.
MS (ES-) m/z = 415 (found for C21H19F3N4O2-H+).
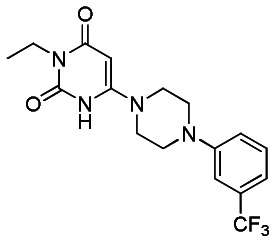
Data for SR-1395 (Probe Analog)
SR-03000001395
SR-03000001395 (97% last step, purity >98%) obtained as a white powder, was synthesized following the same procedures described for SR-01000621848, replacing at the first stage cyclohexylurea by ethylurea and at the last step 1-(3-chlorophenyl) piperazine by 1-(3-(trifluoromethyl)phenyl)piperazine.
FTIR: 2972, 1718, 1695, 1604, 1585, 1496, 1445, 1311, 1231, 1151, 1122, 1097, 1075, 956, 809, 787, 767, 700 cm−1.
1H NMR (400 MHz, (CDCl3)): 1.23 (t, J = 6.9 Hz, 3H), 3.33–3.38 (m, 4H), 3.50–3.56 (m, 4H), 3.95 (q, J = 7.1 Hz, 2H), 5.03 (bs, 1H), 7.10 (dd, J = 8.4, 2.5 Hz, 1H ), 7.12–7.15 (m, 1H), 7.17 (d, J = 7.7 Hz, 1H), 7.41 (d, J = 8.0 Hz, 1H), 10.43 (s, 1H)
13C NMR (100 MHz, (CDCl3)): 13.1, 35.4, 46.2, 48.3, 80.1, 112.8 (q, J = 3.7 Hz), 117.2 (q, J = 3.8 Hz), 119.3, 124.0 (q, J = 272.7 Hz), 129.9, 131.7 (q, J = 31.8 Hz), 150.5, 153.1, 153.3, 163.4.
MS (ES-) m/z = 367 (found for C17H19F3N4O2-H+).

Figure 3Synthesis of SR-1393 (Probe Analog)
Data for 6-chloro-3-isobutylpyrimidine-2,4(1H,3H)-dione, 1H NMR (400 MHz, (CDCl3)): 0.92 (d, J = 6.8 Hz, 6H), 2.12 (n, J = 6.8 Hz, 1H), 3.74 (d, J = 7.5 Hz, 2H), 5.86 (s, 1H), 9.41 (bs, 1H).
SR-03000001393 (97% last step, purity >98%) obtained as a white powder, was synthesized following the same procedures described for SR-01000621848, replacing at the first stage cyclohexylurea by isobutylurea and at the last step 1-(3-chlorophenyl)piperazine by 1-(3-(trifluoromethyl)phenyl)piperazine.
FTIR: 2959, 1698, 1621, 1494, 1447, 1310, 1229, 1165, 1119, 1098, 1075, 994, 950, 785, 696 cm−1.
1H NMR (400 MHz, (CDCl3)): 0.94 (d, J = 6.8 Hz, 6H), 2.15 (n, J = 6.8 Hz, 1H), 3.32–3.39 (m, 4H), 3.50–3.56 (m, 4H), 3.73 (d, J = 7.5 Hz, 2H), 5.02 (bs, 1H), 7.10 (dd, J = 8.4, 2.5 Hz, 1H ), 7.12–7.15 (m, 1H), 7.17 (d, J = 7.7 Hz, 1H), 7.41 (d, J = 8.0 Hz, 1H), 10.53 (s, 1H).
13C NMR (100 MHz, (CDCl3)): 20.2, 27.2, 46.2, 47.2, 48.3, 80.0, 112.60 (q, J = 3.8 Hz), 117.1 (q, J = 3.8 Hz), 119.3, 124.1 (q, J = 272.9 Hz), 129.9, 131.8 (q, J = 32.0 Hz), 150.6, 153.3, 153.6, 163.9
MS (ES-) m/z = 395 (found for C19H23F3N4O2-H+).
3. Results
3.1. Summary of Screening Results
This LRH1 probe development effort was a center-based initiative. The MLPCN collection was not screened.
3.2. Dose Response Curves for Probes and Aromatase Assay

Figure 4Dose response curves for SR-01000621848 (Probe 1, ML180) and SR-01000001309 (Probe 2, ML179) two LRH-1 modulator probes
293T cells were cotransfected with (A and C) full length LRH-1 and Cyp19 aromatase reporter, or (B and D) full length SF-1 and 5xSFRE reporter and treated with various concentrations of either SR-01000621848 (A and B) or SR-03000001309 (C and D) for 20 hours prior to luciferase activity measurement. Relative change was determined by normalizing to vehicle treatment. Treatment with both probes, SR-01000621848 (Max Rep=52% ; IC50=3.7μM) and SR-03000001309 (Max Rep = 46% ; IC50 = 320nM) showed selective efficacy against LRH-1 over SF-1.
These aromatase dose response data are available in PubChem as AID 488782.
3.3. Scaffold/Moiety Chemical Liabilities
There are no known chemical liabilities associated with the probes ML180 and ML179, or the identified probe analogs.
Discussion of SAR Studies
Starting with the initial selective LRH1 inverse agonist hit, SR-1848 (CID 3238389; ML180), we performed an extensive SAR study via analog purchase and synthesis. Three different sites in SR-1848 have been modified. A range of substituted aryl and heteroaryl groups have been introduced in position R1 (see Table 4 in Section 3.4). In general, aryl units at the R1 position possessing meta substituents (e.g., meta-Cl as in SR-1848; a CF3 group at this position in SR-1309, or the substituted quinoline in SR-1174) result in the greatest LRH1 repression. The optimal substituents at the R2 position—which give rise to the lowest IC50 values for LRH1 inverse agonism, are alkyl groups like isobutyl in SR-1393 or cyclohexyl in many other analogs (e.g., SR-1309), or ethyl in SR-135. Introduction of a substituent at R3 leads to a significant reduction or elimination of LRH1 activity; the results obtained thus far indicate that R3 = H is required.
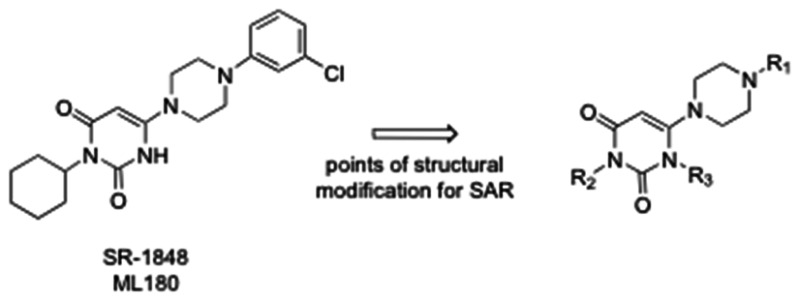
Figure 5Points of Structural Modification for SAR
Table 2SAR of Probes and Selected Analogs
| Structure | Identification Numbers | LRH1 IC50 (μM) | LRH1 Max Repression |
|---|---|---|---|
 | CID 3238389 (ML180, Probe 1) SR-1848 | 3.7 | 64% (10 μM) |
 | CID 45100448 (ML179, Probe 2) SR-1309 | 0.281 | 40% (550 nM) |
 | CID 45382271 (probe analog) SR-1393 | 0.061 | 35% (200 nM) |
 | CID 45480137 (probe analog) SR-1409 | 0.65 | 24% (2 μM) |
 | CID 45382284 (probe analog) SR-1395 | 0.378 | 40% (5 μM) |
 | CID 45100455 (probe analog) SR-1310 | 0.285 | 42% (5 μM) |
 | CID 44825223 (probe analog) SR-1174 | 4 | 75% (10 μM) |
We selected SR-1848 (ML180) and SR-1309 (ML179) as the probes because these two compounds give the best results in inhibition of cancer cell growth. While SR-1848 is only 3.7 μM IC50 vs. LRH1, it gives the maximum LRH1 repression of all compounds studied to date (64% at 10 μM). Probe ML179 (SR-1309) is 13-fold more potent (IC50) than ML180 (SR-1848) against LRH1, but demonstrates a lower maximum repression of LRH1 transcription compared to ML180 (40% vs. 64%), albeit at a lower concentration. While SR-1393 (CID 45382271) is the most potent LRH1 inverse agonist discovered to date (IC50 = 61 nM), it is weaker in repressing LRH1 transcription (35% at 200 nM) than SR-1309. In tests of these compounds against various cancer cell lines, SR-1309 proved superior to SR-1393 in the majority of cell lines studied, and therefore SR-1309 was declared a probe rather than SR-1393.
3.4. SAR Tables
Table 3SAR of Probe Compounds
| Compound Information | LRH1 %INH (AID 485348) | LRH1 Dose Response (AID 488782) | SF1 Fold Change (AID 488779) | SF1 Dose Response (AID 488780) | VP16 Fold Change (AID 488775) | QPCR mRNA Fold Change (AID 488769) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample ID | Structure | Source | MLS ID | SID | CID | LRH-1 AVG % Inhibition (at 5 μM) | SD | LRH-1 IC50 | SD | SF-1 Avg Fold Change (at 5 μM) | SD | SF-1 IC50 | SD | VP-16 Avg Fold Change (at 5μM) | SD | Haptoglob in mRNA Avg Fold change (at 10 μM) | SD | SAA1 mRNA Avg Fold change (at 10 μM) | SD | SAA4 mRNA Avg Fold change (at 10 μM) | SD |
| PROBE #1 SR-01000621848-2 |
 | P | MLS003 153119 | 99344023 | 3238389 | 52.33% (Active) | 0.055 | 3.7μM (Active) | 0.031 | 4.67% (Inactive) | 0.13 | > 10 μM (Inactive) | 0.067 | 1.3% (Inactive) | 0.012 | 0.87 (Active) | 0.04 | 0.91 (Active) | 0.00 | 0.55 (Active) | 0.09 |
| PROBE #2 SR-03000001309-1 |
 | S | MLS003 153122 | 92092843 | 45100448 | 45.67% (Active) | 0.051 | 320nM (Active) | 0.075 | −1% | 0.01 | > 10 μM (Inactive) | −6% | 0.049 | 0.86 (Active) | 0.04 | 0.93 (Active) | 0.04 | 0.55 (Active) | 0.10 | |
Analogs with “ND” indicates that their activities for a particular anti-target or mechanism-of-action assay were Not Determined due to lack of LRH1 activity compared to probes. Only the two probes were tested in the LRH1 target gene QPCR assay.
Table 4SAR of R1 Analogs
| Compound Information: R1 Analogs | LRH1 %INH (AID 485348) | LRH1 Dose Response (AID 488782) | SF1 Fold Change (AID 488779) | SF1 Dose Response (AID 488780) | VP16 Fold Change (AID 488775) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample ID | Structure | Source | MLS ID | SID | CID | LRH-1 AVG %Inhibition (at 5 μM) | SD | LRH-1 IC50 | SD | SF-1 Avg Fold Change (at 5 μM) | SD | SF-1 IC50 | SD | VP-16 Avg Fold Change (at 5μM) | SD |
| SR-03000001170-1 |
 | S | 89649733 | 44825215 | 35.33% (Active) | 0.045 | 921nM (Active) | 0.069 | 9.33% (Inactive) | 0.08 | 9.2μM (Inactive) | 0.06 | 0% | 0.000 | |
| SR-03000001307-1 |
 | P | 92092833 | 1484049 | 45% (Active) | 0.026 | 538nM (Active) | 0.065 | −3% | 0.06 | N/D | −7% | 0.058 | ||
| SR-03000001310-1 |
 | S | MLS003153123 | 92092844 | 45100455 | 42% (Active) | 0.147 | 430nM (Active) | 0.100 | 3.3% (Inactive) | 0.08 | N/D | −5% | 0.050 | |
| SR-01000140561-3 |
 | P | 89650166 | 3244825 | 32% (Active) | 0.087 | 2.6μM (Active) | 0.087 | −3% | 0.06 | N/D | −16% | 0.124 | ||
| SR-03000001171-1 |
 | S | 89649734 | 44825216 | 42% (Active) | 0.053 | 4.8μM (Active) | 0.038 | 0.67% (Inactive) | 0.11 | N/D | −27% | 0.058 | ||
| SR-03000001174-1 |
 | S | MLS003153125 | 89649737 | 44825223 | 34.667% (Active) | 0.042 | 4.0μM (Active) | 0.242 | 11.33% (Inactive) | 0.04 | N/D | 7% (Inactive) | 0.026 | |
| SR-03000001302-1 |
 | S | 92092825 | 45100433 | 32.33% (Active) | 0.045 | ND | 7% (Inactive) | 0.07 | ND | 17% (Active) | 0.044 | |||
| SR-01000621845-2 |
 | P | 89650164 | 3240644 | 1.33% (Inactive) | 0.121 | ND | −2% | 0.11 | ND | −9% | 0.017 | |||
| SR-03000001172-1 |
 | S | 89649735 | 44825230 | 36% (Active) | 0.036 | 20μM (Inactive) | 0.017 | −7% | 0.08 | N/D | −8% | 0.076 | ||
| SR-03000001173-1 |
 | S | 89649736 | 44825240 | 30.33% (Active) | 0.074 | 80μM (Inactive) | 0.027 | −4% | 0.05 | N/D | −4% | 0.066 | ||
| SR-01000140537-3 |
 | P | 92092838 | 3241826 | 18% (Inactive) | 0.156 | ND | 6% | 0.11 | ND | −12% | 0.072 | |||
| SR-03000001383-1 |
 | S | 93375297 | 45382280 | 16.33% (Inactive) | 0.035 | ND | −12% | 0.12 | ND | 0% | 0.006 | |||
| SR-03000001384-1 |
 | S | 93375298 | 45382275 | 38.333% (Active) | 0.040 | 13μM (Inactive) | 0.015 | −4% | 0.13 | N/D | N/D | −6% | 0.085 | |
| SR-03000001385-1 |
 | S | 93375299 | 45382282 | 16% (Inactive) | 0.095 | ND | 5% (Inactive) | 0.09 | ND | 14.3% (Active) | 0.021 | |||
| SR-03000001386-1 |
 | S | 93375300 | 45382276 | 36.67% (Active) | 0.091 | ND | −112% | 0.19 | ND | ND | ||||
| SR-03000001387-1 |
 | S | 93375301 | 45382268 | 20.3% (Inactive) | 0.091 | ND | −83% | 0.44 | ND | −12% | 0.030 | |||
| SR-03000001388-1 |
 | S | 93375302 | 45382285 | 16% (Inactive) | 0.090 | ND | 13.33% (Inactive) | 0.10 | ND | 16% (Active) | 0.090 | |||
| SR-03000001389-1 |
 | S | 93375303 | 45382283 | 20% (Inactive) | 0.046 | ND | −9% | 0.07 | ND | 1.3% (Inactive) | 0.012 | |||
| SR-01000140541-4 |
 | P | 92092841 | 3571734 | 5.33% (Inactive) | 0.055 | ND | 4.33% (Inactive) | 0.06 | ND | 0% | 0.100 | |||
| SR-03000001175-1 |
 | S | 89649738 | 44825217 | 0% (Inactive) | 0.121 | ND | 1% (Inactive) | 0.06 | ND | −5% | 0.084 | |||
| SR-03000001176-1 |
 | S | 89649739 | 44825219 | 2.67% (Inactive) | 0.081 | ND | −14% | 0.17 | ND | −4% | 0.105 | |||
| SR-03000001300-1 |
 | S | 92092823 | 45100453 | −0.30% | 0.173 | ND | −13% | 0.21 | ND | −48% | 0.157 | |||
| SR-03000001301-1 |
 | S | 92092824 | 45100462 | 16.67% (Inactive) | 0.055 | ND | 23.67% (Active) | 0.08 | ND | −5% | 0.031 | |||
| SR-01000607332-2 |
 | S | 92092826 | 3237172 | 11% (Inactive) | 0.046 | ND | 6% (Inactive) | 0.05 | ND | 16.67% (Active) | 0.235 | |||
| SR-03000001390-1 |
 | S | 93375304 | 45382269 | 3.33% (Inactive) | 0.042 | ND | −49% | 0.01 | ND | 26% (Active) | 0.020 | |||
| SR-03000001391-1 |
 | S | 93375305 | 45382272 | 11% | 0.144 | ND | 5.33% (Inactive) | 0.13 | ND | 13.3% (Active) | 0.050 | |||
| SR-03000001403-1 |
 | S | 93577916 | 45480135 | 16% (Inactive) | 0.050 | ND | −19% | 0.06 | ND | 1.3% (Inactive) | 0.257 | |||
| SR-03000001404-1 |
 | S | 93577917 | 45480133 | 17% (Inactive) | 0.053 | ND | −23% | 0.06 | ND | −4% | 0.035 | |||
| SR-03000001410-1 |
 | S | 93577923 | 45480139 | 3.333% (Inactive) | 0.035 | ND | 9% (Inactive) | 0.04 | ND | 2.67% (Inactive) | 0.032 | |||
| SR-01000140539-2 |
 | P | 85786762 | 1479244 | 1.667% (Inactive) | 0.012 | ND | 4.67% (Inactive) | 0.02 | ND | 1.33% (Inactive) | 0.015 | |||
| SR-03000001303-1 |
 | S | 92092827 | 45100460 | 13.33% (Inactive) | 0.115 | ND | 8.67% (Inactive) | 0.10 | ND | 26% (Active) | 0.131 | |||
| SR-03000001304-1 |
 | S | 92092828 | 45100430 | 4% (Inactive) | 0.139 | ND | −8% | 0.03 | ND | 11.33% (Inactive) | 0.045 | |||
| SR-03000001305-1 |
 | S | 92092829 | 45100439 | 0% | 0.006 | ND | 1.67% (Inactive) | 0.08 | ND | 8% (Inactive) | 0.072 | |||
| SR-01000621846-2 |
 | P | 92092839 | 3239085 | 2.33% (Inactive) | 0.045 | ND | 9% (Inactive) | 0.05 | ND | 26.3% (Active) | 0.110 | |||
| SR-01000140511-3 |
 | P | 92092840 | 3245522 | 16% (Inactive) | 0.183 | ND | 19.3% (Inactive) | 0.14 | ND | −6% | 0.053 | |||
| SR-01000140541-4 |
 | P | 92092841 | 3571734 | 5.33% (Inactive) | 0.061 | ND | 5.3% (Inactive) | 0.08 | ND | 0% | 0.100 | |||
| SR-01000101040-3 |
 | P | 92092842 | 3571735 | 10% (Inactive) | 0.020 | ND | −5% | 0.07 | ND | 5.67% (Inactive) | 0.060 | |||
| SR-01000140507-3 |
 | S | 92092831 | 4141058 | 20.33% (Inactive) | 0.101 | ND | 23% (Inactive) | 0.13 | ND | 0% | 0.006 | |||
| SR-03000001306-1 |
 | S | 92092830 | 45100432 | 8.67% (Inactive) | 0.075 | ND | 5% (Inactive) | 0.17 | ND | −6% | 0.053 | |||
| SR-01000101020-3 |
 | S | 92092832 | 3242688 | 5.33% (Inactive) | 0.136 | ND | 0% | 0.01 | ND | 9% (Inactive) | 0.052 | |||
| SR-03000001311-1 |
 | S | 92092845 | 45100438 | 14.33% (Inactive) | 0.127 | ND | 23% | 0.08 | ND | −11% | 0.032 | |||
ND indicates a compound was not tested in the indicated assay due to lack of LRH1 activity compared to probe in the 3X%INH assay
Table 5SAR of R2 Analogs
| Compound Information: R2 Analogs (with variable R1) | LRH1 %INH (AID 485348) | LRH1 Dose Response (AID 488782) | SF1 Fold Change (AID 488779) | SF1 Dose Response (AID 488780) | VP16 Fold Change (AID 488775) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample ID | Structure | Source | MLS ID | SID | CID | LRH-1 AVG %Inhibition (at 5 μM) | SD | LRH-1 IC50 | SD | SF-1 Avg Fold Change (at 5 μM) | SD | SF-1 IC50 | SD | VP-16 Avg Fold Change (at 5μM) | SD |
| SR-03000001393-1 |
 | S | MLS003153124 | 93375307 | 45382271 | 33.33% (Active) | 0.098 | 110nM (Active) | 0.023 | −95% | 0.15 | 15μM (Inactive) | 0.02 | 8.33% (Inactive) | 0.015 |
| SR-01000097977-3 |
 | P | 92092835 | 5310579 | 22.67% (Inactive) | 0.140 | ND | −18% | 0.05 | ND | −5% | 0.064 | |||
| SR-01000008560-2 |
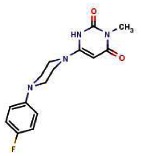 | P | 85786757 | 7080816 | 14% (Inactive) | 0.122 | ND | −2% | 0.07 | ND | −13% | 0.095 | |||
| SR-01000008566-2 |
 | P | 85786758 | 7080823 | 40.67% (Active) | 0.116 | 1.1μM (Active) | 0.023 | −2% | 0.07 | N/D | −1% | 0.091 | ||
| SR-01000101016-2 |
 | P | 85786759 | 7080833 | 45% (Active) | 0.176 | 4.2μM (Active) | 0.036 | −10% | 0.10 | N/D | 4.67% (Inactive) | 0.031 | ||
| SR-03000001394-1 |
 | S | 93375308 | 45382279 | 36.67% (Active) | 0.012 | 570nM (Active) | 0.052 | −164% | 0.21 | 5μM (Inactive) | 0.03 | 1% (Inactive) | 0.017 | |
| SR-03000001395-1 |
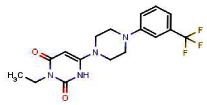 | S | MLS003 153121 | 93375309 | 45382284 | 39.33% (Active) | 0.049 | 1μM Active) | 0.183 | −81% | 0.01 | N/D | 1.3% (Inactive) | 0.045 | |
| SR-03000001409-1 |
 | S | MLS003 153120 | 93577922 | 45480137 | 17.67% (Inactive) | 0.040 | ND | −3% | 0.10 | ND | −3% | 0.148 | ||
| SR-03000001392-1 |
 | S | 93375306 | 45382277 | 3% (Inactive) | 0.010 | ND | 1.3% (Inactive) | 0.09 | ND | 6% (Inactive) | 0.046 | |||
| SR-01000101326-2 |
 | P | 85786761 | 7080827 | 2.33% (Inactive) | 0.076 | ND | 10.3% (Inactive) | 0.05 | ND | −7% | 0.040 | |||
| SR-01000101018-2 |
 | P | 85786760 | 6623965 | 3.67% (Inactive) | 0.035 | ND | 7% (Inactive) | 0.03 | ND | −7% | 0.095 | |||
| SR-03000001149-1 |
 | S | 89649715 | 44825226 | 0% | 0.173 | ND | 2% (Inactive) | 0.02 | ND | 2.67% (Inactive) | 0.031 | |||
| SR-03000001308-1 |
 | P | 92092834 | 4621936 | 5.667% (Inactive) | 0.136 | ND | 0% | 0.08 | ND | −7% | 0.030 | |||
| SR-01000101008-3 |
 | P | 92092836 | 7080829 | 9.67% (Inactive) | 0.087 | ND | 26.67% (Active) | 0.11 | ND | −10% | 0.100 | |||
| SR-01000769836-2 |
 | P | 92092837 | 7080831 | 3.33% (Inactive) | 0.049 | ND | 30% (Active) | 0.04 | ND | 0% | 0.006 | |||
| SR-03000001405-1 |
 | S | 93577918 | 45480136 | 21.3% (Inactive) | 0.047 | ND | 3% (Inactive) | 0.06 | ND | −1% | 0.206 | |||
| SR-03000001406-1 |
 | S | 93577919 | 45480138 | 29.67% (Active) | 0.025 | ND | −24% | 0.12 | ND | 20.33% (Active) | 0.307 | |||
| SR-03000001407-1 |
 | S | 93577920 | 45480134 | 2% (Inactive) | 0.020 | ND | 3.67% (Inactive) | 0.04 | ND | 2% (Inactive) | 0.062 | |||
ND indicates a compound was not tested in the indicated assay due to lack of LRH1 activity compared to probe in the 3X%INH assay
3.5. Cellular Activity
The probe is active in a variety of cell-based assays performed by Dr. Griffin. Importantly, these assays demonstrate that Probe #1 (ML180) and Probe #2 (ML179) are potent in a variety of breast cancer cell lines. These MTT data are available in PubChem as AID 504928.
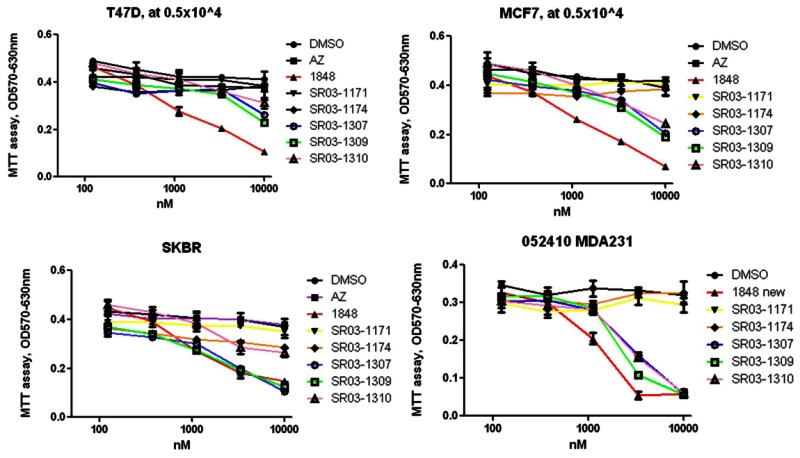
Figure 6Probes #1 ML180 (1848) and #2 ML179 (SR03-1309) Potency in 4 Different Breast Cancer Cell Lines (Dose Response Curves)
Function Assays: Star Reporter Assays
LRH1 inverse agonist probe compounds (powder samples) were also tested in assays to monitor their effects on the promoter activity of the Steroidogenic acute regulatory protein (Star). These data are available in PubChem as AID 504933. The dose response curves for each probe are below, in Figure 7.

3.6. Profiling Assays
Selectivity of the probe compounds over the nuclear receptor SF-1 has been monitored and is presented in the previous sections. ML180 did not modulate GAL4-VP16. In addition, probe 1 (ML180; 1848) has been tested against a library of all 48 human nuclear receptors [43]. Preliminary analysis suggests that ML180 has little activity on other nuclear receptors (see Figure 8). Note that SR-1848 (ML180) is not active on LRH1 in this assay. We have confirmed that ML180 is active only on full length LRH1 and inactive on the Gal4-LBD-LRH1 construct used in the assay d. These data can also be found in PubChem as AID 504934.

Figure 8
ML180 Nuclear Receptor Promiscuity.
ML180 (MLS003153119 = CID 3238389) was tested in 470 assays in PubChem and was active in 15 AIDs (3.19%). Eleven of these are protein targets with five of the 11 being cytochrome P450’s. With this knowledge we will closely monitor P450 activity of ML180 analogs. Two other targets were RORA (actual hit is in the SF-1 counterscreen to RORA) and the SF-1 assay. This is the origin of the probes as SF-1 is the closest family member to LRH1. However, as we show above, while ML180 is active on GAL4-SF1, it is not active on full length receptor. The remaining four targets are DNA damage-inducible transcript 3, E3 ubiquitin-protein ligase Mdm2, Mdm4, and centromeric isoform d. Activity is these assays could be indirect and LRH1 dependent.
ML180 was also active in; 1) Luminescence Cell-Based Primary HTS to Identify Inhibitors of Beta Cell Apoptosis. [Primary Screening], 2) Luminescence Cell-Based Dose Retest to Confirm Inhibitors of Beta Cell Apoptosis [Confirmatory], 3) uHTS luminescence assay for the identification of chemical inhibitors of B-cell specific antigen receptor-induced NF-kB activation [Primary Screening], and 4) HTS to identify inhibitors of zVAD Induced Cell Death in L929 Cells [Primary Screening].
We are confident that ML180 is not an inhibitor of luciferase as we see selectivity over SF-1 in a luminescence-based luciferase reporter assay.
4. Discussion
4.1. Comparison to existing art and how the new probe is an improvement
Currently there are no published inverse agonists of LRH1.
4.2. Mechanism of Action Studies
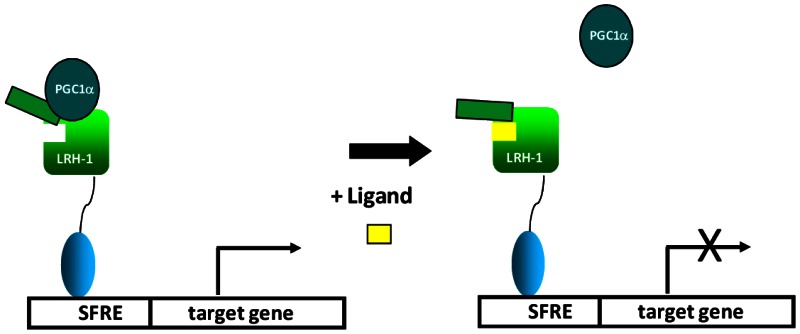
Figure 9AProbe MOA (Step 1)
LRH1 is a constitutively active nuclear receptor that binds to identical response elements as SF-1 in promoter regions of LRH1 target genes. The scheme above suggests that LRH1 is associated with NR coactivators such as PGC1a and this interaction drives transactivation of LRH1 target genes. Upon binding to LRH1, inverse agonists such as ML180 alter the conformational dynamics of the receptor causing dissociation of co-activator. It is also likely that inverse agonists enhance LRH1 interaction with NR co-repressors such as SHP.
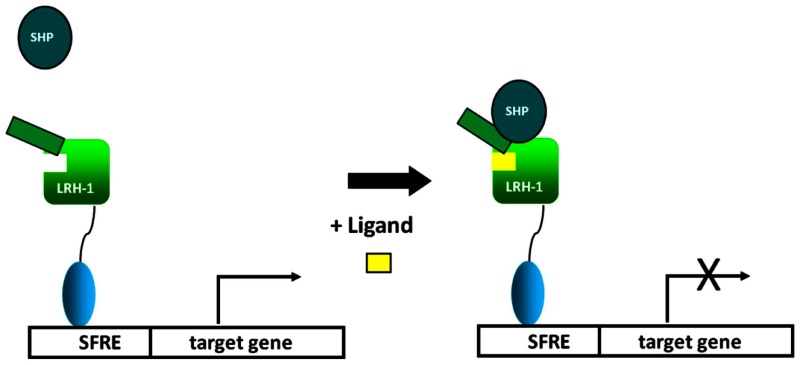
Figure 9BProbe MOA (Step 2)

Figure 9CProbe MOA (Step 3)
Acute Phase Response (APR)- Preliminary studies in our lab using Huh7 cells stimulated with IL1β and IL6 results in significant increase in expression of LRH1, haptoglobin (Hp), serum amyloid A-4 (SAA1), and serum amyloid A-4 (SAA4) as determined by qPCR (see PubChem AID 488769). Surprisingly and opposite of the anticipated outcome, treatment of these cells with the inverse agonists ML180 and ML179 represses the expression of three of these genes (Hp, SAA1, SAA4) to the level of control cells (unstimulated) and LRH1 to levels below that in control cells. This finding confirms that our inverse agonists can repress the expression of LRH1 and more importantly, suggests that LRH1 inverse agonists can repress APR genes in similar fashion to LRH1 agonists. Clearly the mechanism of action of inverse agonists in this model is likely different than that of agonists. This finding warrants further mechanistic studies which will be carried out as part of this extended probe development project.

Figure 10Hp, SAA1 & SAA4 Expression in the Presence and Absence of Probes
The above QPCR data have been submitted to PubChem as AID 488769.
LRH1 Modulation of Aromatase Expression – preliminary studies in our lab show that treatment of hepG2 cells with SR-1848 reduces the expression of aromatase cytochrome p450 gene (Cyp19). Treatment of these cells with a siRNA for LRH1 also reduces the expression of aromatase. Treatment of cells with siRNA for LRH1 blunts the effects of SR-1848 (probe ML180) on aromatase expression. These results have been submitted to PubChem as AID 488782. Optimization of Cyp19 knockdown is ongoing. The relevance of these studies is found in prior discovery that aromatase is highly upregulated in breast cancers as well as breast tissue surrounding tumors.

4.3. Planned Future Studies
We intend to continue to explore the SAR in this LRH1 inverse agonist series, in order to identify selective LRH1 probes that have IC50’s <100 nM and >50% maximal repression of LRH1 expression at ≤200 nM. The PK properties of the lead compounds will also be optimized using a series of in vitro and in vivo (mouse) PK studies to identify a compound that is suitable for use in animal studies. In addition to the assays described above, advanced compounds will be tested in the following:
HTRF cell-based aromatase activity assay: LRH1 has been shown to control the expression of Cyp19 (aromatase) and as such inverse agonists should reduce the level of aromatase activity in breast cancer cells. To test this hypothesis, we will use a cell-based HTRF aromatase assay as described here. This method is a homogeneous assay for monitoring the aromatase-mediated conversion of estrone into estradiol in breast cancer cells. The assay is a competitive immunoassay in which estradiol produced by breast cancer cells (e.g. ER-positive MCF7 or ER-negative MDA-MB-231) and XL665-labeled estradiol compete for binding to an Anti-estradiol MAb labeled with Cryptate. Due to the proximity between the Cryptate and the XL665 molecules, maximum FRET (Fluorescence Resonance Energy Transfer) is generated when the sample does not contain any estradiol. The signal decreases with increasing estradiol concentration present in the breast cancer cell supernatant. The assay has been developed by CisBio and will be run as follows. Briefly, cells (12000–25000 cells/well in 100 μL in 96-well plate) are grown in culture media supplemented with 10% charcoal-depleted FBS for 48h at 37 °C. The cells are washed with PBS and 50 μL of culture media containing various concentrations of the test compound of interest are added to the corresponding wells followed by further incubation for 24h at 37 °C. After this incubation step, the optimal estrone concentration is added to the wells and further incubated for 24h at 37 °C. At the detection step, 10 μL of the cell supernatant are mixed with 5 uL of Anti-estradiol Cryptate conjugate and 5 μL of Estradiol-XL665 and incubated for 2h at RT in 384-well plates (black). Fluorescence signal can be read for instance on a Viewlux ultraHTS microplate imager from PerkinElmer.
Invasion Assay: Compounds that are potent inverse agonists of LRH1 and that are active in the MTT assay will be tested in an invasion assay. We will use the BD BioCoat Tumor Invasion System which is ideal for use to test prospective anti-metastatic compounds. This system is suitable for automation and is compatible with most fluorescence plate readers and liquid handling devices. The plates contain a uniform layer of matrigel matrix which acts as a reconstituted basement membrane providing a barrier to non-invasive cells but providing an appropriate protein structure to study invasion. The coating process occludes the pores of the membrane, blocking non-invasive cells from migrating through the membrane. In contrast, invasive cells (malignant and non-malignant cells) are able to detach themselves from and migrate through the coated membrane. The assay will be run as recommended by the manufacturer.
in vitro metabolism studies: in vitro metabolic stability studies will be performed by the DMPK laboratory at Scripps Florida, to predict the relative rate of biotransformation to help select candidates that are likely to have favorable pharmacokinetics in vivo. Drug candidates are incubated (separately) with pooled human, mouse and rat microsomes and cofactors to determine the rate of metabolism. Incubations will be performed using a 96-well format to increase sample throughput. Typical drug concentrations studied are 1 μM and 10 μM, providing concentrations that are comparable to plasma drug levels that are efficacious. Samples will be collected at multiple time points and the concentration of the parent drug will be determined using HPLC coupled to a triple quadrupole mass spectrometer (LC/MS-MS), allowing the calculation of half-life for each drug. In select cases, we may also proceed with further LC/MS-MS analysis of samples to structurally identify the metabolites. In cases where the most promising compounds have poor metabolic stability, medicinal chemistry will be applied to modify the regions of the compounds undergoing metabolism. Compounds will also be assessed for their (in)ability to inhibit cytochrome P450’s. We desire lead compounds that have IC50’s ≥ 10 μM against a battery of six major human CYP’s that are run on a weekly basis at Scripps Florida.
in vivo mouse PK: In vivo pharmacokinetics of selected compounds (up to 5 per year) will be assessed in C57Bl6 mice (n = 24). Compounds are dosed intravenously at 1 mg/kg of orally by gavage at 2 mg/kg. Blood is taken at eight time points and collected into EDTA containing tubes and plasma was generated using standard centrifugation techniques. Because of the small blood volume in mice, each mouse is bled only two times. Plasma proteins are precipitated with acetonitrile and drug concentrations are determined by LC-MS/MS. Data are fit by WinNonLin using a noncompartmental model and basic pharmacokinetic parameters including peak plasma concentration (Cmax), oral bioavailability, exposure (AUC), half life (t1/2), clearance (CL), and volume of distribution (Vd) are calculated. The concentration of drug in the brain will also be measured. All procedures are approved by the Scripps Florida IACUC and the Scripps Florida vivarium is fully AAALAC accredited.
Tumor xenograft in athymic mice: The purpose of these experiments is to study LRH1 inverse agonists for their ability to treat breast cancers, using the standard breast cancer xenograft model in nude mice. This is one of the best studied tumor biology systems for assessing the effects of drugs such as SERMs and aromatase inhibitors, as tumors induced by MCF-7 and T47D breast cancer cells are highly sensitive to hormonal treatment, and also eventually develop acquired resistance as seen in patients. Thus this is an excellent model to test the effects of LRH1 inverse agonists as these compounds should impact levels of estrogen in these animals. Preliminary results suggest that our probe compounds ML179 and ML180 are effective at reducing the proliferation of ER-negative breast cancer cells thus we will also include the use of MDA-MB-231 cells in this model. Briefly, athymic mice will be injected subcutaneously in the lower back with a suspension of MCF-7, T47D, or MDA-MB-231 human breast cancer cells (~106 cells) in sterile saline with Matrigel. Compound treatment (10 mg/kg intraperitoneally) and vehicle only controls typically begins approximately 2–4 weeks after implantation when tumors reach ~500 cubic millimeters in size.
DIO mouse model: 4 to 5 week old C57BL/6J mice will be obtained from The Jackson Laboratory. DIO animals (6 animals per group) will be fed a high fat diet for 3 months (Clinton/Cybulsky rodent diet, 40% kcal from fat, devoid of cholesterol; Research Diets, New Brunswick NJ). Lean control mice will receive a standard diet during the same time period. For glucose tolerance tests, mice will be intraperitoneally (i.p.) injected with 10mg/kg of the LRH1 modulator or vehicle only for 6 days, and fasted overnight before i.p. injection of 2 g/kg D-glucose. Blood samples will be collected at time 0, and 15, 30, 60 and 120 min after injection of the glucose Plasma glucose, triglycerides and total lipids will be measured using standard techniques. Body weights and food intake will also be measured.
5. References
- 1.
- Clyne CD, Speed CJ, Zhou J, Simpson ER. Liver Receptor Homologue-1 (LRH-1) Regulates Expression of Aromatase in Preadipocytes. J. Biol. Chem. 2002;277(23):20591–20597. [PubMed: 11927588]
- 2.
- Zhao Y, Agarwal VR, Mendelson CR, Simpson ER. Estrogen biosynthesis proximal to a breast tumor is stimulated by PGE2 via cyclic AMP, leading to activation of promoter II of the CYP19 (aromatase) gene. Endocrinology. 1996;137(12):5739–42. [PubMed: 8940410]
- 3.
- Harada N. Aberrant expression of aromatase in breast cancer tissues. J Steroid Biochem Mol Biol. 1997;61(3–6):175–84. [PubMed: 9365188]
- 4.
- Agarwal VR, Bulun SE, Leitch M, Rohrich R, Simpson ER. Use of alternative promoters to express the aromatase cytochrome P450 (CYP19) gene in breast adipose tissues of cancer-free and breast cancer patients. J Clin Endocrinol Metab. 1996;81(11):3843–9. [PubMed: 8923826]
- 5.
- Bouchard MF, Taniguchi H, Viger RS. Protein Kinase A-Dependent Synergism between GATA Factors and the Nuclear Receptor, Liver Receptor Homolog-1, Regulates Human Aromatase (CYP19) PII Promoter Activity in Breast Cancer Cells. Endocrinology. 2005;146(11):4905–4916. [PubMed: 16109788]
- 6.
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, Maloney PR, Willson TM, Kliewer SA. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6(3):517–26. [PubMed: 11030332]
- 7.
- Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. Molecular Basis for Feedback Regulation of Bile Acid Synthesis by Nuclear Receptors. Molecular Cell. 2000;6(3):507–515. [PubMed: 11030331]
- 8.
- Bulun SE, Price TM, Aitken J, Mahendroo MS, Simpson ER. A link between breast cancer and local estrogen biosynthesis suggested by quantification of breast adipose tissue aromatase cytochrome P450 transcripts using competitive polymerase chain reaction after reverse transcription. J Clin Endocrinol Metab. 1993;77(6):1622–8. [PubMed: 8117355]
- 9.
- Clyne CD, Kovacic A, Speed CJ, Zhou J, Pezzi V, Simpson ER. Regulation of aromatase expression by the nuclear receptor LRH-1 in adipose tissue. Molecular and Cellular Endocrinology. 2004;215(1–2):39–44. [PubMed: 15026173]
- 10.
- Schoonjans K, Dubuquoy L, Mebis J, Fayard E, Wendling O, Haby C, Geboes K, Auwerx J. Liver receptor homolog 1 contributes to intestinal tumor formation through effects on cell cycle and inflammation. Proceedings of the National Academy of Sciences. 2005;102(6):2058–2062. [PMC free article: PMC548586] [PubMed: 15684064]
- 11.
- Chand AL, Herridge KA, Thompson EW, Clyne CD. The orphan nuclear receptor LRH-1 promotes breast cancer motility and invasion. Endocr Relat Cancer. 2010;17(4):965–75. [PubMed: 20817789]
- 12.
- Fayard E, Auwerx J, Schoonjans K. LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol. 2004;14(5):250–60. [PubMed: 15130581]
- 13.
- Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77(4):481–90. [PubMed: 8187173]
- 14.
- Li Y, Lambert MH, Xu HE. Activation of Nuclear Receptors: A Perspective from Structural Genomics. Structure. 2003;11(7):741–746. [PubMed: 12842037]
- 15.
- Ueda H, Sun GC, Murata T, Hirose S. A novel DNA-binding motif abuts the zinc finger domain of insect nuclear hormone receptor FTZ-F1 and mouse embryonal long terminal repeat-binding protein. Mol Cell Biol. 1992;12(12):5667–72. [PMC free article: PMC360506] [PubMed: 1448096]
- 16.
- Li Y, Choi M, Cavey G, Daugherty J, Suino K, Kovach A, Bingham NC, Kliewer SA, Xu HE. Crystallographic identification and functional characterization of phospholipids as ligands for the orphan nuclear receptor steroidogenic factor-1. Mol Cell. 2005;17(4):491–502. [PubMed: 15721253]
- 17.
- Solomon IH, Hager JM, Safi R, McDonnell DP, Redinbo MR, Ortlund EA. Crystal structure of the human LRH-1 DBD-DNA complex reveals Ftz-F1 domain positioning is required for receptor activity. J Mol Biol. 2005;354(5):1091–102. [PubMed: 16289203]
- 18.
- Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, Juzumiene D, Bynum JM, Madauss K, Montana V, Lebedeva L, Suzawa M, Williams JD, Williams SP, Guy RK, Thornton JW, Fletterick RJ, Willson TM, Ingraham HA. Structural Analyses Reveal Phosphatidyl Inositols as Ligands for the NR5 Orphan Receptors SF-1 and LRH-1. Cell. 2005;120(3):343–355. [PubMed: 15707893]
- 19.
- Whitby RJ, Dixon S, Maloney PR, Delerive P, Goodwin BJ, Parks DJ, Willson TM. Identification of Small Molecule Agonists of the Orphan Nuclear Receptors Liver Receptor Homolog-1 and Steroidogenic Factor-1. J. Med. Chem. 2006;49(23):6652–6655. [PubMed: 17154495]
- 20.
- Madoux F, Li X, Chase P, Zastrow G, Cameron MD, Conkright JJ, Griffin PR, Thacher S, Hodder P. Potent, selective and cell penetrant inhibitors of SF-1 by functional ultra-high-throughput screening. Mol Pharmacol. 2008;73(6):1776–84. [PMC free article: PMC3228235] [PubMed: 18334597]
- 21.
- Wang W, Zhang C, Marimuthu A, Krupka HI, Tabrizizad M, Shelloe R, Mehra U, Eng K, Nguyen H, Settachatgul C, Powell B, Milburn MV, West BL. The crystal structures of human steroidogenic factor-1 and liver receptor homologue-1. Proceedings of the National Academy of Sciences. 2005;102(21):7505–7510. [PMC free article: PMC1140416] [PubMed: 15897460]
- 22.
- Li Y, Choi M, Suino K, Kovach A, Daugherty J, Kliewer SA, Xu HE. Structural and biochemical basis for selective repression of the orphan nuclear receptor liver receptor homolog 1 by small heterodimer partner. Proceedings of the National Academy of Sciences. 2005;102(27):9505–9510. [PMC free article: PMC1157103] [PubMed: 15976031]
- 23.
- Francis GA, Fayard E, Picard F, Auwerx J. Nuclear receptors and the control of metabolism. Annu Rev Physiol. 2003;65:261–311. [PubMed: 12518001]
- 24.
- Nitta M, Ku S, Brown C, Okamoto AY, Shan B. CPF: an orphan nuclear receptor that regulates liver-specific expression of the human cholesterol 7alpha-hydroxylase gene. Proc Natl Acad Sci USA. 1999;96(12):6660–5. [PMC free article: PMC21971] [PubMed: 10359768]
- 25.
- del Castillo-Olivares A, Gil G. Alpha 1-fetoprotein transcription factor is required for the expression of sterol 12alpha -hydroxylase, the specific enzyme for cholic acid synthesis. Potential role in the bile acid-mediated regulation of gene transcription. J Biol Chem. 2000;275(23):17793–9. [PubMed: 10747975]
- 26.
- Luo Y, Liang CP, Tall AR. The orphan nuclear receptor LRH-1 potentiates the sterol-mediated induction of the human CETP gene by liver X receptor. J Biol Chem. 2001;276(27):24767–73. [PubMed: 11331284]
- 27.
- Delerive P, Galardi CM, Bisi JE, Nicodeme E, Goodwin B. Identification of Liver Receptor Homolog-1 as a Novel Regulator of Apolipoprotein AI Gene Transcription. Mol Endocrinol. 2004;18(10):2378–2387. [PubMed: 15218078]
- 28.
- Schoonjans K, Annicotte JS, Huby T, Botrugno OA, Fayard E, Ueda Y, Chapman J, Auwerx J. Liver receptor homolog 1 controls the expression of the scavenger receptor class B type I. EMBO Rep. 2002;3(12):1181–7. [PMC free article: PMC1308324] [PubMed: 12446566]
- 29.
- Venteclef N, Smith JC, Goodwin B, Delerive P. Liver Receptor Homolog 1 Is a Negative Regulator of the Hepatic Acute-Phase Response. Mol. Cell. Biol. 2006;26(18):6799–6807. [PMC free article: PMC1592867] [PubMed: 16943422]
- 30.
- Venteclef N, Delerive P. Interleukin-1 Receptor Antagonist Induction as an Additional Mechanism for Liver Receptor Homolog-1 to Negatively Regulate the Hepatic Acute Phase Response. J. Biol. Chem. 2007;282(7):4393–4399. [PubMed: 17158876]
- 31.
- Venteclef N, Jakobsson T, Ehrlund A, Damdimopoulos A, Mikkonen L, Ellis E, Nilsson LM, Parini P, Janne OA, Gustafsson JA, Steffensen KR, Treuter E. GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRbeta in the hepatic acute phase response. Genes Dev. 2010;24(4):381–95. [PMC free article: PMC2816737] [PubMed: 20159957]
- 32.
- Galarneau L, Pare JF, Allard D, Hamel D, Levesque L, Tugwood JD, Green S, Belanger L. The alpha1-fetoprotein locus is activated by a nuclear receptor of the Drosophila FTZ-F1 family. Mol Cell Biol. 1996;16(7):3853–65. [PMC free article: PMC231382] [PubMed: 8668203]
- 33.
- Kudo T, Sutou S. Molecular cloning of chicken FTZ-F1-related orphan receptors. Gene. 1997;197(1–2):261–8. [PubMed: 9332374]
- 34.
- Boerboom D, Pilon N, Behdjani R, Silversides DW, Sirois J. Expression and regulation of transcripts encoding two members of the NR5A nuclear receptor subfamily of orphan nuclear receptors, steroidogenic factor-1 and NR5A2, in equine ovarian cells during the ovulatory process. Endocrinology. 2000;141(12):4647–56. [PubMed: 11108279]
- 35.
- Broadus J, McCabe JR, Endrizzi B, Thummel CS, Woodard CT. The Drosophila beta FTZ-F1 orphan nuclear receptor provides competence for stage-specific responses to the steroid hormone ecdysone. Mol Cell. 1999;3(2):143–9. [PubMed: 10078197]
- 36.
- Ellinger-Ziegelbauer H, Hihi AK, Laudet V, Keller H, Wahli W, Dreyer C. FTZ-F1-related orphan receptors in Xenopus laevis: transcriptional regulators differentially expressed during early embryogenesis. Mol Cell Biol. 1994;14(4):2786–97. [PMC free article: PMC358644] [PubMed: 8139576]
- 37.
- Lavorgna G, Ueda H, Clos J, Wu C. FTZ-F1, a steroid hormone receptor-like protein implicated in the activation of fushi tarazu. Science. 1991;252(5007):848–51. [PubMed: 1709303]
- 38.
- Chand A, Herridge KA, Thompson EW, Clyne C. The orphan nuclear receptor LRH-1 promotes breast cancer motility and invasion. Endocr Relat Cancer. 2010 [PubMed: 20817789]
- 39.
- Li X, He Y, Ruiz CH, Koenig M, Cameron MD, Vojkovsky T. Characterization of dasatinib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways. Drug Metab Dispos. 2009;37(6):1242–50. [PMC free article: PMC3202349] [PubMed: 19282395]
- 40.
- Li X, Kamenecka TM, Cameron MD. Bioactivation of the epidermal growth factor receptor inhibitor gefitinib: implications for pulmonary and hepatic toxicities. Chem Res Toxicol. 2009;22(10):1736–42. [PubMed: 19803472]
- 41.
- Devi IB, PJ An expedient method for the synthesis of 6-substituted uracils under microwave irradiation in a solvent-free medium. Tetrahedron Lett. 2005;46(34):5727–5729.
- 42.
- Zhi C, Long ZY, Gambino J, Xu WC, Brown NC, Barnes M, Butler M, LaMarr W, Wright GE. Synthesis of substituted 6-anilinouracils and their inhibition of DNA polymerase IIIC and Gram-positive bacterial growth. J Med Chem. 2003;46(13):2731–9. [PubMed: 12801236]
- 43.
- Kumar N, Solt LA, Conkright JJ, Wang Y, Istrate MA, Busby SA, Garcia-Ordonez RD, Burris TP, Griffin PR. The benzenesulfoamide T0901317 [N-(2,2,2-trifluoroethyl)-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl]-benzenesulfonamide] is a novel retinoic acid receptor-related orphan receptor-alpha/gamma inverse agonist. Mol Pharmacol. 2010;77(2):228–36. [PMC free article: PMC2812071] [PubMed: 19887649]
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- Calcineurin and CRTC2 mediate FSH and TGFβ1 upregulation of Cyp19a1 and Nr5a in ovary granulosa cells.[J Mol Endocrinol. 2014]Calcineurin and CRTC2 mediate FSH and TGFβ1 upregulation of Cyp19a1 and Nr5a in ovary granulosa cells.Lai WA, Yeh YT, Fang WL, Wu LS, Harada N, Wang PH, Ke FC, Lee WL, Hwang JJ. J Mol Endocrinol. 2014 Oct; 53(2):259-70. Epub 2014 Jul 23.
- The orphan nuclear receptor LRH-1 promotes breast cancer motility and invasion.[Endocr Relat Cancer. 2010]The orphan nuclear receptor LRH-1 promotes breast cancer motility and invasion.Chand AL, Herridge KA, Thompson EW, Clyne CD. Endocr Relat Cancer. 2010 Dec; 17(4):965-75. Epub 2010 Oct 29.
- Divergent sequence tunes ligand sensitivity in phospholipid-regulated hormone receptors.[J Biol Chem. 2013]Divergent sequence tunes ligand sensitivity in phospholipid-regulated hormone receptors.Musille PM, Pathak M, Lauer JL, Griffin PR, Ortlund EA. J Biol Chem. 2013 Jul 12; 288(28):20702-12. Epub 2013 Jun 4.
- Review LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis.[Trends Cell Biol. 2004]Review LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis.Fayard E, Auwerx J, Schoonjans K. Trends Cell Biol. 2004 May; 14(5):250-60.
- Review Campaign to identify novel modulators of the Retinoic acid receptor-related Orphan Receptors (ROR).[Probe Reports from the NIH Mol...]Review Campaign to identify novel modulators of the Retinoic acid receptor-related Orphan Receptors (ROR).Kumar N, Solt LA, Conkright J, Wang Y, Istrate MA, Busby SA, Garcia-Ordonez RD, Nuhant P, Burris T, Mercer BA, et al. Probe Reports from the NIH Molecular Libraries Program. 2010
- Discovery of Inverse Agonists for the Liver Receptor Homologue-1 (LRH1; NR5A2) -...Discovery of Inverse Agonists for the Liver Receptor Homologue-1 (LRH1; NR5A2) - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...