

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

BLM helicase is a DNA unwinding enzyme critical in DNA repair via the homologous recombination (HR) pathway. Mutations of the BLM gene result in diminished BLM helicase activity and can cause Bloom’s Syndrome, which is characterized by a long list of phenotypes, including predisposition to cancers. Similar to other DNA repair enzymes, inhibition of BLM helicase results in sensitization of tumor cells to conventional cancer therapies, such as camptothecin. Currently, there are no known small molecule inhibitors of BLM helicase; thus, the discovery of a novel small molecule inhibitor would help define the mechanism of action of the enzyme and provide a basis for future development of inhibitors and cancer therapeutics. The first-in-class probe molecule described herein (ML216) displays low micromolar potency and selectivity over related helicases, such as RECQ1, RECQ5, and E. coli UvrD helicases. This probe also inhibits cell proliferation of BLM-proficient fibroblast cells (PSNF5) while having only minimal effects on BLM-deficient fibroblast cells (PSNG13), indicating on-target activity in a cellular context. In addition, the probe molecule increases the frequency of sister chromatid exchanges, a diagnostic cellular phenotype consistent with the absence of a functional BLM protein.
Assigned Assay Grant #: MH087284
Screening Center Name & PI: NIH Chemical Genomics Center, Christopher Austin
Chemistry Center Name & PI: NIH Chemical Genomics Center, Christopher Austin
Assay Submitter & Institution: Ian Hickson & Opher Gileadi, Structural Genomics Consortium, University of Oxford, UK
PubChem Summary Bioassay Identifier (AID): 2386
Probe Structure & Characteristics
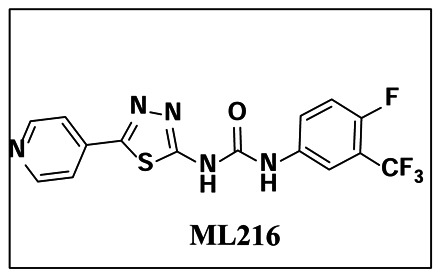
| CID/ML# | Target Name | IC50/EC50 (μM) [SID, AID] | Anti-target Name(s) | IC50/EC50 (μM) [SID, AID] | Fold Selective | Secondary Assay(s) Name:
IC50/EC50 (μM) [SID, AID] |
|---|---|---|---|---|---|---|
| CID 49852229/ML216 | BLM Helicase | DNA unwinding radio-labeled gel-based
assay: 1.8 μM [SID 104222021, AID 504666] | RecQ1 | ~50 μM | ~28-fold | DNA unwinding, fluorescent gel-based
assay: 1.21 ± 0.53 μM [SID 104222021, AID 504662] |
| RecQ5 | >50 μM | >28-fold | ||||
| UvrD helicases | >50 μM | >28-fold selective | ||||
| [SID 104222021, AID 504663] |
Recommendations for Scientific Use of the Probe
ML216 is the first selective BLM Helicase small molecule probe that inhibits with low micromolar potency. Its lack of activity against related helicases RECQ1, RECQ5, and UvrD (in E. coli) makes ML216 ideally suitable for use in helicase research and the broader field of DNA repair research. The anti-proliferative activity of the probe in BLM-proficient fibroblast cells (PSNF5) and its increase of frequency in sister chromatid exchanges enhances the utility of the probe in cellular studies. Lastly, ML216 is a suitable starting point for further mouse tumor xenograft models and for the development of potential small molecule cancer therapeutics.
1. Introduction
The goal of this project is to develop small molecule inhibitors of BLM helicase activity that are selective over related RECQ family members and can be used to examine the consequences of inhibition on survival of cancer cells. The importance of a chemical probe is enhanced by the fact that there are currently no published reports of a non-promiscuous BLM helicase inhibitor.
The human genome, which is under constant mutagenic threat, relies heavily on complicated mechanisms of detecting and repairing DNA in order to propagate. One such process is carried out by the RECQ family helicase, BLM. Although the precise function of BLM helicase still remains unclear, it has been proposed that it promotes genome stability through the homologous recombination (HR) repair pathway.1,2 In humans, BLM helicase binds directly to topoisomerase III. The protein has unwinding activity of the G-quadruplex structure and is believed to be a player in telomere maintenance.3 Additionally, BLM helicase unwinds Holliday junctions and interacts with a key recombinase protein, Rad51, suggesting an important role in recombinatorial DNA repair.2,3
Specific mutation of the BLM gene results in a rare genetic disorder known as Bloom’s syndrome, which is characterized by small physical stature, immunodeficiency, and a profound predisposition to cancers.4,5 Although Bloom’s syndrome patients are rare, individuals that possess a BLM gene mutation may be faced with a higher probability of developing breast cancer6 and colorectal cancer,7,8 among other cancers. Treatment of Bloom’s syndrome remains a difficult task, but targeting the downstream symptoms of cancer is a significant and attainable goal. BLM helicase responds to DNA damage by accumulating rapidly at sites of laser-induced double strand breaks or sites of γ-irradiation, further illustrating its role in DNA repair and targeting BLM as a potential cancer treatment target.9 While the biological pathways of BLM helicase have been the focus of much research, the dearth of small-molecule inhibitors of this enzyme has hampered further elucidation of its biological importance. Consequently, it has been our goal to develop a small molecule probe to assist in deciphering the exact mechanism of the enzyme and its role in DNA repair.
Non-specific DNA binding ligands, such as Distamycin A and camptothecin, have been used to inhibit both BLM and a closely related helicase, WRN, in the micromolar range.10 Subsequent reports of substituted acridines described potent inhibitors of the telomerase enzyme complex, which in turn were also highly potent non-specific inhibitors of RECQ family helicases.11 These functions of BLM prompted recent discoveries of siRNAs that induce apoptosis in cancer cells by suppressing the activity of the BLM gene and other RECQ helicases.3,12 However, while these studies help validate BLM as a therapeutic target, they do not account for any selective non-DNA binding small molecule inhibitors, which is the goal of this project.
The targeting of specific DNA repair proteins and their respective pathways to enhance the DNA damaging effects of current chemotherapy has been proposed as an attractive anticancer strategy. This combined synergistic therapy is based on the principle of synthetic lethality, which represents an approach that takes advantage of the intrinsic DNA repair deficiencies of specific tumor cells. Specifically, BLM-deficient cells have shown sensitivity to the known cancer chemotherapeutic drugs camptothecin, cis-platin, hydroxyurea, and 5-fluorouracil13 (see Figure 1). The concept of targeting inhibitors of the DNA repair pathway has been recently validated by the success of several small molecule inhibitors of PARP (Poly ADP ribose polymerase) in clinical trials.14 PARP inhibitors are particularly effective in people with the BRCA1 and BRCA2 mutations, which are prevalent in many prostate, breast and ovarian cancers. While patients with these mutations are typically predisposed to developing a wide variety of cancers, these cancer cells are particularly sensitive to PARP inhibitors due to synthetic lethality as described above. Thus, the identification of small molecules that specifically inhibit BLM helicase may assist in uncovering the synthetic lethal relationships between BLM and other DNA repair pathways and lead to the development of novel combination anticancer regimens.
Figure 1
Known DNA damaging agents and the recently reported WRN helicase inhibitor, NSC19630.
Due to the lack of any small molecule inhibitors of BLM helicase, the current effort focuses on a selective inhibitor of BLM helicase. A recent screen for small molecule inhibitors of WRN helicase yielded a selective agent that also potentiated anticancer drug treatment of sensitized cells.15 This report represents the growing interest in the field of small molecule inhibitors for DNA repair enzymes, specifically helicases. The small molecule BLM inhibitors described herein provide structure-activity relationships, potency, selectivity, and activity in cell-based systems.
Prior Art
There are no known specific inhibitors of BLM helicase, although recent work has been done to inhibit a related helicase, WRN.15 This compound is part of the NCI diversity set, and is likely a promiscuous and reactive molecule (see Figure 1). The growing interest in modulators of DNA repair enzymes makes BLM helicase an important and interesting target. Distamycin A and camptothecin are two examples of non-specific micromolar inhibitors of both BLM and WRN (Figure 1).
2. Materials and Methods
General Methods for Chemistry. Unless otherwise stated, all reactions were carried out under an atmosphere of dry argon or nitrogen in dried glassware. Indicated reaction temperatures refer to those of the reaction bath, while room temperature (rt) is noted as 25°C. All solvents were of anhydrous quality, purchased from Aldrich Chemical Co. and used as received. Commercially available starting materials and reagents were purchased from Aldrich and were used as received.
Analytical thin layer chromatography (TLC) was performed with Sigma Aldrich TLC plates (5 × 20 cm, 60 Å, 250 μm). Visualization was accomplished by irradiation under a 254 nm UV lamp. Chromatography on silica gel was performed using forced flow (liquid) of the indicated solvent system on Biotage KP-Sil pre-packed cartridges and the Biotage SP-1 automated chromatography system. 1H- and 13C NMR spectra were recorded on a Varian Inova 400 MHz spectrometer. Chemical shifts are reported in ppm with the solvent resonance as the internal standard (CDCl3 7.26 ppm, 77.00 ppm, DMSO-d6 2.49 ppm, 39.51 ppm for 1H, 13C, respectively). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), coupling constants, and number of protons. Low resolution mass spectra (electrospray ionization) were acquired on an Agilent Technologies 6130 Quadrupole spectrometer coupled to the HPLC system. High resolution mass spectral data was collected in-house using an Agilent 6210 time-of-flight mass spectrometer, also coupled to an Agilent Technologies 1200 series HPLC system. If needed, products were purified via a Waters semi-preparative HPLC equipped with a Phenomenex Luna® C18 reverse phase (5 micron, 30 × 75 mm) column having a flow rate of 45 ml/min. The mobile phase was a mixture of acetonitrile (0.025% TFA) and H2O (0.05% TFA), and the temperature was maintained at 50°C.
Samples were analyzed for purity on an Agilent 1200 series LC/MS equipped with a Luna® C18 reverse phase (3 micron, 3 × 75 mm) column having a flow rate of 0.8–1.0 ml/min over a 7-minute gradient and an 8.5 minute run time. Purity of final compounds was determined to be >95%, using a 3 μL injection with quantitation by AUC at 220 and 254 nm (Agilent Diode Array Detector).
2.1. Assays
BLM qHTS assay
Inhibition of BLM activity was screened by utilizing a fork duplex DNA substrate [20-bp duplex with 30-nt oligo(dT) tails], which harbored a rhodamine-type fluorophore (TAMRA) at the 3′-end (see Table 1, A1) and a non-fluorescent Black Hole Quencher-2 (BHQ-2) at the opposing 5′-end (see Table 1, A2). An increase in the fluorescence intensity as a result of the ATP-dependent unwinding of the duplex region by BLM was used to measure the enzyme activity. The screening protocol was as follows: 3 μL of BLM (10 nM) in a reaction buffer containing 50 mM Tris-HCl (pH 8.0), 5 mM NaCl, 2 mM MgCl2, 1 mM DTT, 0.01% Tween-20, and 2.5 μg/ml poly(dI-dC) were dispensed to 1536-well Greiner black solid bottom plates. Compounds (23 nl) were transferred to the plates via Kalypsys pintool and were incubated for 15 min at room temperature. Then 1 μL of substrate solution (200 nM fork duplex and 2 mM ATP) was added to start the reaction. The plates were immediately transferred into ViewLux High-throughput CCD imager (Perkin-Elmer) in order to measure the reaction progress in a discontinuous kinetic mode (two reads spaced one hour apart, with intermediate room temperature incubation of the covered assay plate) using 525 nm excitation and 598 nm emission fluorescence protocol. The fluorescence intensity difference between the two reads was used to compute reaction progress. Ellagic acid (CID 5281855) was used as an interplate control at a two-fold, 16-point dilution in duplicate to produce final concentrations in the 5.75 μM to 0.175 nM range.
Table 1
Oligonucleotide substrates.
Preparation of radiolabeled oligonucleotide BLM substrates
To prepare the radiolabeled fork duplex substrate, A3 (see Table 1) was 5′-end labeled using T4 polynucleotide kinase and [γ-32P] ATP and then annealed with the unlabeled complementary strand A4 (see Table 1) by heating to 100°C and slowly cooling to room temperature. Similarly, the Holliday junction substrate was prepared by annealing the 5′-end labeled A3 oligonucleotide with A4, A5, and A6 (see Table 1). The annealed oligonucleotides were gel-purified and dialyzed against buffer containing 10 mM Tris-HCl (pH 7.5) and 10 mM MgCl2. The concentrations of the radiolabeled substrates were determined using scintillation counting (Beckman LS 5000CE).
DNA unwinding measured by gel electrophoresis (32P-labeled substrate)
Initial screening hits were validated by the standard assay for measuring DNA helicase activity, which utilizes gel electrophoresis to monitor the helicase-mediated unwinding of a radiolabeled DNA substrate. Four unwinding assays using a number of DNA helicases (BLM, UvrD, RecQL5, and RecQL1) were carried out in a reaction buffer containing 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 2 mM MgCl2, 2 mM ATP, 1 mM DTT, and 0.1 mg/ml BSA. Optimal substrate and enzyme concentrations, as well as reaction times, were determined experimentally. All reactions were performed at 37°C and terminated by the addition of loading buffer containing 20 mM EDTA and 2.5% bromophenol blue. Reaction products were separated on 15% nondenaturing polyacrylamide gels in 1× TBE at 30 mA for 1 hr at 4°C. The gels were then dried and visualized by autoradiography (Kodak BioMax MR-1).
DNA unwinding measured by gel electrophoresis (fluorescent-labeled substrate)
A gel-based assay employing a fluorescent fork duplex substrate was also utilized to ascertain SAR of select inhibitors. The fork duplex substrate was prepared by annealing the 3′-TAMRA-labeled A1 oligonucleotide (see Table 1) with the unlabeled complementary strand A2 (see Table 1). The standard reaction contained 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 2 mM MgCl2, 1 mM DTT, 0.1% Tween-20, and 5 nM BLM. Serial dilutions of selected inhibitors were added at a final concentration of 5% DMSO. The reactions were initiated by addition of 2 mM ATP and 200 nM TAMRA-labeled fork duplex substrate. Following incubation at room temperature for 30 min, the reactions were terminated by the addition of loading dye containing 5% glycerol, 2 mM EDTA, and 2.5% bromophenol blue. Reaction products were then separated on 12% nondenaturing polyacrylamide gels in 1× TAE and visualized using a Bio-Rad ChemiDocTM XRS Gel Imager.
Fluorescence polarization assay
Equilibrium binding of DNA helicases (i.e. BLM and RecQ1) to TAMRA-labeled oligonucleotides was monitored by fluorescence polarization in 384-well plate format (Greiner, black solid bottom). Serial dilutions of selected inhibitors were mixed with the indicated amounts of DNA helicase and TAMRA-labeled oligonucleotides in a binding buffer containing 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 2 mM MgCl2, 1 mM DTT, and 0.1% Tween-20. Following incubation at room temperature for ~2 hr, plates were read on the ViewLux High-throughput CCD imager (Perkin-Elmer) using the polarization filter set with excitation and emission wavelengths of 525 and 598 nm, respectively.
Cell proliferation assay
The BLM-deficient (PSNG13) and BLM-proficient (PSNF5) fibroblast cells were seeded into separate wells of a 96-well plate. The cells were grown overnight and then treated with BLM inhibitors at the concentration described for 72 hrs. WST-1 reagent (Roche Applied Science, Burgess Hill, UK) was added at a 1:10 dilution for 4 hours at 37°C in 5% CO2 as per the manufacturer’s instructions. The 96-well plates were then analyzed optically at 450 nm with a reference wavelength of 690 nm using a Bio-Rad Benchmark microplate reader (Bio-Rad). See Section 3.5 (Figure 5) for results.
Sister chromatid exchange assay
PSNG13 or PSNF5 cells were incubated with medium containing 10 μg/ml BrdU for 36 hours, followed by treatment with BLM inhibitors or DMSO for a further 36 hours. 100 ng/ml KaryoMAX Colcemid (Gibco, Paisley, UK) was added for the final 30 min of the treatment period. One hundred metaphase nuclei were scored for each analysis using a Nikon Eclipse 80i microscope equipped with Lucia G software (Laboratory Imaging, Prague, Czech Republic). Differences in the distribution of SCEs were compared using a paired t-test. See Section 3.5 (Figure 6) for results.
2.2. Probe Chemical Characterization

*Purity >95% as judged by LC/MS and 1H NMR
1-(4-fluoro-3-(trifluoromethyl)phenyl)-3-(5-(pyridin-4-yl)-1,3,4-thiadiazol-2-yl)urea: LC-MS: rt (min) = 3.13; 1H NMR (400 MHz, DMSO-d6) δ 9.45–9.65 (m, 1H), 8.69–8.77 (m, 2H), 8.02–8.11 (m, 1H), 7.83–7.91 (m, 2H), 7.70–7.82 (m, 1H) and 7.49 (s, 1H); HRMS (ESI) m/z (M+H)+ calcd. for C15H10F4N5OS, 384.0551; found 384.0549.
LC/MS conditions
- LC/MS (Agilent system) Retention time t1 (short) = 3.13 min and t2 (long) = 4.49 min.
- Column: 3 × 75 mm Luna C18, 3 micron
- Run time: 4.5 min (short); 8.5 min (long)
- Gradient: 4 % to 100 %
- Mobile phase: Acetonitrile (0.025 % TFA), water (0.05 % TFA).
- Flow rate: 0.8 to 1.0 ml
- Temperature: 50 °C
- UV wavelength: 220 nm, 254 nm
MLS Numbers for probe analogs
| MLS IDs | NCGC IDs | SID | CID | |
|---|---|---|---|---|
| MLS003370591 | NCGC00189393-01 | 104222021 | 49852229 | ML216/Compound 17 |
| MLS003370592 | NCGC00187635-01 | 104221977 | 1075698 | Analog |
| MLS003370593 | NCGC00189397-01 | 104222027 | 49852237 | Analog |
| MLS003370594 | NCGC00241737-01 | 104224676 | 49853190 | Analog |
| MLS003370595 | NCGC00241739-01 | 104224678 | 49853281 | Analog |
| MLS003370596 | NCGC00187638-01 | 104221979 | 49852226 | Analog |

Figure 2Stability of ML216 in DPBS buffer at room temperature over 48 hrs
Percent remaining after 48 hours = 100%.
Synthetic route to ML216
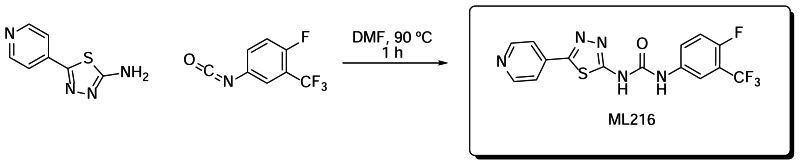
Scheme 1Preparation of ML216
2.3. Probe Preparation
Preparation of 1-(4-fluoro-3-(trifluoromethyl)phenyl)-3-(5-(pyridin-4-yl)-1,3,4-thiadiazol-2-yl)urea: A mixture of 5-(pyridin-4-yl)-1,3,4-thiadiazol-2-amine (0.10 g, 0.56 mmol) and 1-fluoro-4-isocyanato-2-(trifluoromethyl)benzene (0.07 ml, 0.47 mmol) was dissolved in DMF (2 ml) and stirred at 90ºC for 1 hour. The solution was allowed to cool and was quenched with water (1 ml). The resulting precipitate was filtered and washed with water (3 ml), ethyl acetate (3 ml) and acetone (3 ml) to yield pure 1-(4-fluoro-3-(trifluoromethyl)phenyl)-3-(5-(pyridin-4-yl)-1,3,4-thiadiazol-2-yl)urea. Yield: 0.083 mg (46%).
3. Results
3.1. Summary of Screening Results
A robust donor/quencher-based assay was miniaturized to a 1536-well format and used to identify small molecule inhibitors of BLM helicase (see 2.1 Assays). Prior to the full-collection screen, the assay was tested and found to perform reproducibly by screening the LOPAC1280 (library of pharmacologically active compounds) in triplicate using a fully-integrated robotic system. The assay was then applied to screen a 355,254-compound library arrayed as seven-point titrations ranging from 57 μM to 3.6 nM (AID: 2528). In total, 3,216 assay plates were screened in one continuous robotic run. Overall, the assay performed well during the entire course of the screen with the Z′ factor remaining consistent at an average of 0.9. In addition, the intra-plate control titration of ellagic acid (CID 5281855) was stable with an average IC50 of 1.9 μM (Figure 3, shown in green).

Figure 3
Summary of BLM screening results. 3D scatter plot of qHTS data showing concentration-response curves for the intra-plate control titration (green), potential inhibitors (dark and light blue), and compounds that gave signal increase (red).
In contrast to traditional HTS, qHTS provides concentration response curves (CRCs) for each compound and allows for calculation of an AC50 value (defined as the half-maximal activity concentration) for each compound in the primary screen. In this screen, the inhibition associated with each well was computed from the alteration in fluorescence intensity over the time-course measurement period, normalized against control wells. Analysis of the CRCs (curve quality and potency) resulted in 698 top inhibitors with full CRCs (Figure 3, shown in dark blue), 1,881 inconclusive inhibitors (Figure 3, shown in light blue), and 1,636 activators (Figure 3, shown in red). Actives represented 1.19% of the chemical library screened, while the remaining compounds were inactive. Compounds were further filtered by using RECQ1 qHTS (AID: 2549) as a counterscreen. Approximately 200 compounds were tested in the DNA unwinding gel electrophoresis assay (32P-labeled substrate, see 2.1 Assays) for orthogonal confirmation, resulting in the identification of the present chemotype. The initial active from the probe chemotype was MLS000559245/CID 1075698 (2). Subsequent analogs of the probe chemotype were tested in the DNA unwinding gel electrophoresis assay using a fluorescent-labeled substrate (see 2.1 Assays). This assay was utilized to generate the IC50 values shown in SAR Tables 2 and 3.
Table 2
BLM inhibition: Representative analogs.
Table 3
BLM inhibition: Representative analogs.
3.2. Dose Response Curves for Probe
The inhibition of DNA unwinding by both truncated and full-length BLM DNA unwinding for the probe (ML216) is displayed in Figure 4.

3.3. Scaffold/Moiety Chemical Liabilities
The main identified liability of this probe is poor aqueous solubility. Chemistry efforts are ongoing to improve this aspect of the molecule; they will be one of the continued goals of our extended characterization proposal as part of the planned further studies. However, the solubility of this molecule was observed to be much improved under assay conditions used for IC50 determination as compared to simply dissolving in PBS (pH 7.4) buffer. These differences likely result from the presence of detergent (Tween-20), which is known to improve the apparent solubility of the compounds.
3.4. SAR Tables
Investigation of the initial BLM hit, CID 1075698, yielded tractable SAR with definite trends (see Tables 2 and 3). The resynthesized version of the original hit (2) had an activity of 1.6 μM in the DNA unwinding fluorescent gel-based assay. The first round of changes surrounded the western pyridine moiety, which is attached at the 2-position of a thiadiazole core. Removal of aromaticity (14, 23–24, 26) eliminated all activity (50% inhibition was not reached at 50 μM concentration), as did changing the pyridine to a substituted or non-substituted phenyl group (8, 13, 15, 29). Ring systems with free amines (42–43) were prepared in an effort to improve solubility, but again, activity was lost. Movement of the pyridine nitrogen was tolerated at the 3-position (32, IC50 = 3.5 μM), but not at the 2-position (16, IC50 = 40 μM). Since potency was minimally affected with the shift to the 3-pyridine, it was decided to continue studies using the initial 4-pyridine structure.
In an effort to improve potency, the next portion of the molecule to be explored was the eastern ring. Changing the initial bis-chlorophenyl ring to the isosteric 2-naphthalene moiety resulted in very little change in potency (1, 3, 8, 12–16). A variety of aromatic substitutions were tried, including mono chlorination (9), bis-fluorination (25), bis-bromination (27), nitriles (31), and trifluoromethyl alkylation (22). These compounds (mono-chloro aside) generally had potency < 20 μM, indicating that a large range of eastern ring modifications were tolerated. Bis-substitution of the ring was also carried out (11, 17–21, 28, 30, 32–40), resulting in several agents with similar activity to the original hit. Compounds containing nitro groups had noticeably poor solubility and were not pursued despite improved potency. Analogs with the 4-fluoro-3-trifluoromethylphenyl eastern ring and urea linker (17, 32) had good potency, and from empirical observations, solubility did not appear to be a problem for these. Since the gel assays used to track SAR were done manually, the manual inspect led to the observation of no compound precipitation up to the highest concentration tested (50 μM), despite the low solubility measurement in PBS buffer.
In an effort to improve aqueous solubility of the lead compound, the urea and the thiadiazole were modified. Changing the urea to the typically more hydrophilic isosteres such as thiourea (5), guanidine (6), and cyanoguanidine (40) showed a range of potencies, none of which were improved significantly over the original hit. Other commonly used isosteres (41, 46–47, 49–50) were also met with limited success. Since desired activity appeared to favor only the urea and thiourea, emphasis was then placed on the thiadiazole core. The first logical change was to convert the thiadiazole to an oxadiazole (3–4, 38); however, this eliminated all activity. Changes to the thiadiazole core, such as a pyridine (44, 51) or to the inactive pyrimidine (45), did not greatly improve solubility.
Despite having discovered compounds with better potencies (35 and 36) in the DNA unwinding gel-based assay after several rounds of SAR, it was determined that 17 (ML216) was the probe compound, based on its potency against both truncated (IC50 = 1.2 ± 0.53 μM) and full-length BLM (IC50 = 3.39 ± 1.45 μM) (see Figure 4) as well as its favorable solubility compared to related analogs. This selection was also based on improved cell-based results over the original “hit” compound (2) (vida infra).
SAR Summary

3.5. Cellular Activity
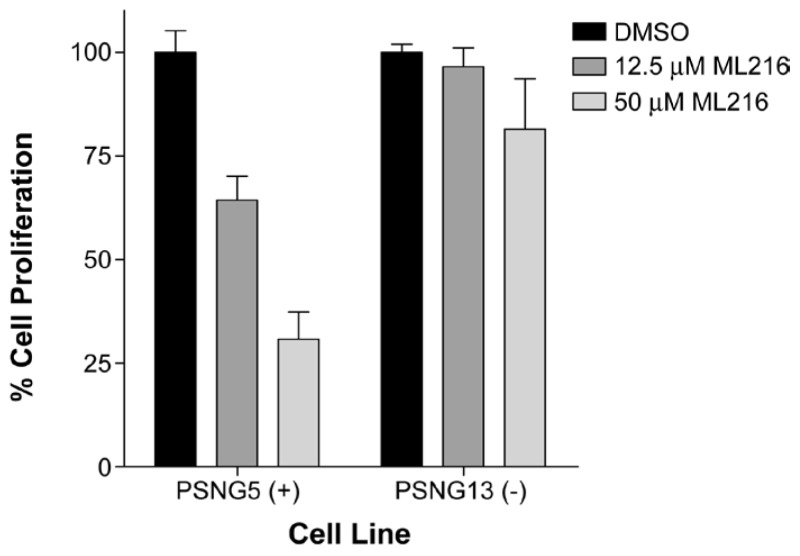
Figure 5Inhibition of cell proliferation of BLM-complemented (PSNF5) fibroblast cell line, but not the BLM-deficient (PSNG13) fibroblast cell line after 72 hr treatment with ML216
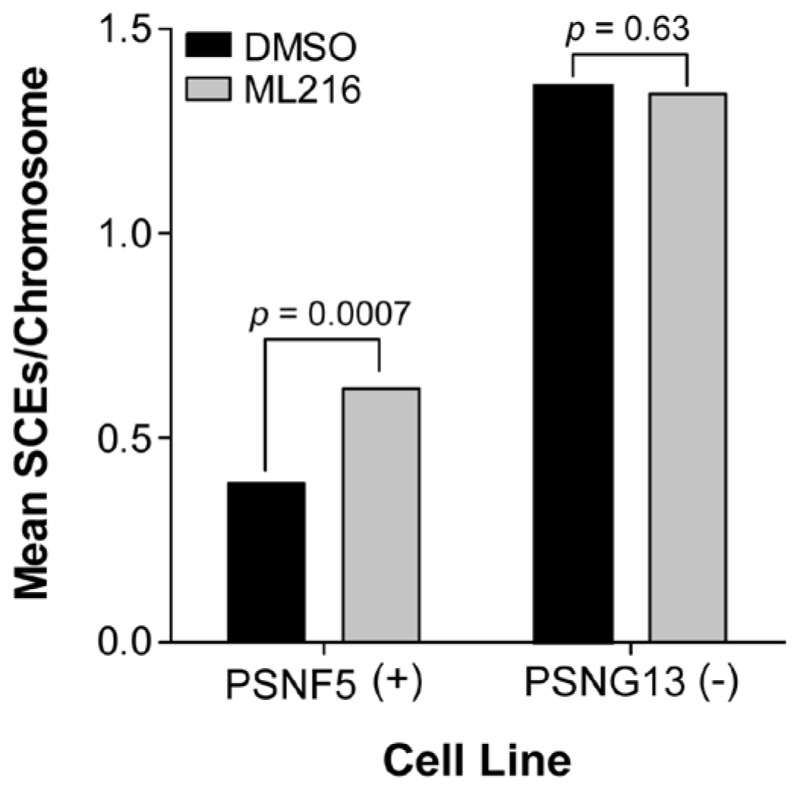
Figure 6Increase in sister chromatid exchanges (SCEs) in the BLM-complemented (PSNF5) fibroblast cell line, but not in the BLM-deficient (PSNG13) fibroblast cell line after treatment with ML216 (50 μM)
3.6. Profiling Assays
Selectivity profiling
Table 4Inhibition selectivity of ML216 on related helicases
| Inhibition at 50 μM compound measured by DNA unwinding assay | ||||
|---|---|---|---|---|
| Compound ID | BLM | UvrD | RecQ1 | RecQ5 |
| ML216 | 98.7% (1.8a)b | 3.9%b | 22.1%c | 11.8%b |
- a
IC50 ( μM) shown in parentheses, if applicable.
- b
Determined by gel electrophoresis (32P-labeled substrate)
- c
Determined by helicase qHTS assay
ADME profiling
Table 5ADME profile of ML216
| Compound | Aq. Kinetic Sol. (PBS @ pH 7.4) | Caco-2 (Papp 10−6 m/s @ pH 7.4) | Efflux Ratio (B→A)/(A→B) | Mouse Liver Microsome Stability (T1/2) | PBS-pH 7.4 Stability:% remaining after 48h | Mouse Plasma Stability:% remaining after 48h |
|---|---|---|---|---|---|---|
| ML216 | 2 μM | 1 | 2 | >120 min | 100 | 100 |
- a
in vitro ADME profile of the probe compound (ML216). Kinetic solubility, Caco-2 permeability, MLM stability and plasma stability were all carried out by Pharmaron Inc. PBS buffer (pH 7.4) stability was performed in-house using LC/MS monitoring UV @ 254 and 220 nm.
4. Discussion
ML216, a first-in-class inhibitor of BLM helicase, was optimized after validating an identified hit from the BLM qHTS of the NIH Molecular Libraries Small Molecule Repository (MLSMR). The probe is selective for BLM (IC50 = 1.2 μM) over related DNA helicases, such as RecQ1 (IC50 ~ 50 μM), RecQ5 (IC50 > 50 μM), and E. coli UvrD (IC50 > 50 μM) helicases (Table 4). Cell-based assays also demonstrate that ML216 selectively inhibits cell proliferation of BLM-proficient fibroblast cells (PSNF5) over BLM-deficient cells (PSNG13) (Figure 5), whereas the initial “hit molecule” 2 shows minimal effect on either cell line. More strikingly, ML216 caused a significant increase in the frequency of sister chromatid exchanges in PSNF5 cells, a key cytogenetic marker of cells lacking BLM activity (Figure 6). Together, these results serve to validate the on-target cellular activity of ML216, as well as highlight a major advantage of the probe molecule over the initial hit. Additionally, ML216 exhibits excellent mouse liver microsomal (MLM) stability and plasma stability (Table 5), indicating that the probe could prove to be useful in vivo. However, for this compound to have appropriate oral bioavailability, additional improvement in the aqueous solubility needs to be made. Current efforts are focused on using the SAR profile outlined in this report to improve this attribute.
4.1. Comparison to Existing Art and How the New Probe is an Improvement
Prior to the discovery of ML216, there were no reported specific small molecules inhibitors of BLM helicase; therefore, this molecule is first-in-class.
4.2. Mechanism of Action Studies
Besides the forked DNA duplex substrate, BLM has been shown to display an ATP-dependent 3′–5′ DNA helicase activity that unwinds a variety of DNA substrates arising during DNA replication and repair such as Holliday junctions, G-quadruplex DNA, and displacement loops (D-loops). The BLM-dependent unwinding of such structures is also inhibited by ML216 (Figure 7), albeit to a lesser extent than the forked DNA duplex substrate. In addition, a fluorescence polarization assay (see 2.1 Assays), which monitored the binding of BLM to a fluorescently-labeled singled-stranded DNA oligonucleotide, was utilized to gain insight into the mechanism of action of ML216. As Figure 8 illustrates, ML216 inhibited the DNA binding of BLM with an IC50 of 5.1 μM, while having only a minimal effect on RecQ1 (IC50 = 101.8 μM). These results suggest the DNA binding domain of BLM as the putative binding pocket for ML216.

Figure 8
Inhibition of DNA binding by fluorescence polarization. A) Schematic diagram of the fluorescence polarization-based helicase-DNA binding assay. B) Inhibition of BLM and RecQ1 DNA binding by ML216.
4.3. Planned Future Studies
Continued chemistry exploration for a more water soluble compound based on ML216 is ongoing. Additionally, investigation of these compounds in cell-based potentiation studies with a variety of DNA damaging cancer chemotherapeutics and use of this compound in mouse tumor xenograft models will be included as part of the extended characterization proposal. These future studies will also involve testing the probe compound and related analogs against the NCI-60 panel to look for cell lines that are hypersensitive to these agents.
5. References
- 1.
- Holloway JK, Morelli MA, Borst PL, Cohen PE. Mammalian BLM helicase is critical for integrating multiple pathways of meiotic recombination. J. Cell Bio. 2010;188:779–789. [PMC free article: PMC2845075] [PubMed: 20308424]
- 2.
- Chu WK, Hanada K, Kanaar R, Hickson ID. BLM has early and late functions in homologous recombination repair in mouse embryonic stem cells. Oncogene. 2010;29:4705–4714. [PubMed: 20531307]
- 3.
- Takagi M, Shimamoto A, Furuichi Y, Sato A. Apoptosis Inducer for Cancer Cell Oct. 2007;182007
- 4.
- Bohr VA. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends in Biochemical Sciences. 2008;33:609–622. [PMC free article: PMC2606042] [PubMed: 18926708]
- 5.
- Ellis NA, Groden J, Ye T-Z, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J. The Bloom’s Syndrome Gene Product Is Homologous to RecQ Helicases. Cell. 1995;83:655–666. [PubMed: 7585968]
- 6.
- Ding S-l, Yu J-C, Chen S-T, Hsu G-C, Kuo S-J, Lin YH, Wu P-E, Shen C-Y. Genetic variants of BLM interact with RAD51 to increase breast cancer susceptibility. Carcinogenesis. 2009;30:43–49. [PubMed: 18974064]
- 7.
- Gruber SB, Ellis NA, Rennert G, Scott KK, Almog R, Kolachana P, Bonner JD, Kirchhoff T, Tomsho LP, Nafa K, Pierce H, Low M, Satagopan J, Rennert H, Huang H, Greenson JK, Groden J, Rapaport B, Shia J, Johnson S, Gregersen PK, Harris CC, Boyd J, Offit K. BLM Heterozygosity and the Risk of Colorectal Cancer. Science. 2002;297:2013. [PubMed: 12242432]
- 8.
- Frank B, Hoffmeister M, Klopp N, Illig T, Chang-Claude J, Brenner H. Colorectal cancer and polymorphisms in DNA repair genes WRN, RMI1 and BLM. Carcinogenesis. 2010;31:442–445. [PubMed: 19945966]
- 9.
- Tikoo S, Sengupta S. Time to Bloom. Genome Integrity. 2010;1:1–7. [PMC free article: PMC2992038] [PubMed: 21050475]
- 10.
- Brosh RM, Karow JK, White EJ, Shaw ND, Hickson ID, Bohr VA. Potent inhibition of Werner and Bloom helicases by DNA minor groove binding drugs. Nucleic Acids Research. 2000;28:2420–2430. [PMC free article: PMC102731] [PubMed: 10871376]
- 11.
- Li J-L, Harrison RJ, Reszka AP, Brosh RM, Bohr VA, Neidle S, Hickson ID. Inhibition of the Bloom’s and Werner’s Syndrome Helicases by G-Quadruplex. Biochemistry. 2001;40:15194–15202. [PubMed: 11735402]
- 12.
- Takagi M, Futami K, Shimamoto A, Furuichi Y. US 2009/0028861A1. Cancer Cell-Specific Apoptosis-Inducing Agents that Target Chromosome Stabilization-Associated Genes. 2009 Jan. 29
- 13.
- Mao FJ, Sidorova JM, Lauper JM, Emond MJ, Monnat RJ. The Human WRN and BLM RecQ Helicases Differentially Regulate Cell Proliferation and Survival after Chemotherapeutic DNA Damage. Cancer Res. 2010;70:6548–6555. [PMC free article: PMC2941797] [PubMed: 20663905]
- 14a.
- Vasiliou S, Castaner R, Bolos J. Olaparib. Drugs of the Future. 2009;34:101.
- 14b.
- Menear KA, Adcock C, Boulter R, Cockcroft XL, Cosey L, Cranston A, Dillon KJ, Drzewiecki J, Garman S, omez S, Javaid H, Kerrigan F, Knights C, Lau A, Loh VM, Matthews IT, Moore S, O’Connor MJ, Smith GC, Martin NM. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: a novel bioavailable inhibitor of polyADP-ribose)polymerase-1. J. Med. Chem. 2008;51:6581–6591. [PubMed: 18800822]
- 15.
- Aggarwal M, Sommers JA, Shoemaker RH, Brosh RM. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. USA. 2011;108:1525–1530. [PMC free article: PMC3029756] [PubMed: 21220316]
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- Synthesis and SAR studies of 5-(pyridin-4-yl)-1,3,4-thiadiazol-2-amine derivatives as potent inhibitors of Bloom helicase.[Bioorg Med Chem Lett. 2013]Synthesis and SAR studies of 5-(pyridin-4-yl)-1,3,4-thiadiazol-2-amine derivatives as potent inhibitors of Bloom helicase.Rosenthal AS, Dexheimer TS, Gileadi O, Nguyen GH, Chu WK, Hickson ID, Jadhav A, Simeonov A, Maloney DJ. Bioorg Med Chem Lett. 2013 Oct 15; 23(20):5660-6. Epub 2013 Aug 13.
- A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells.[Chem Biol. 2013]A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells.Nguyen GH, Dexheimer TS, Rosenthal AS, Chu WK, Singh DK, Mosedale G, Bachrati CZ, Schultz L, Sakurai M, Savitsky P, et al. Chem Biol. 2013 Jan 24; 20(1):55-62.
- Structure of human Bloom's syndrome helicase in complex with ADP and duplex DNA.[Acta Crystallogr D Biol Crysta...]Structure of human Bloom's syndrome helicase in complex with ADP and duplex DNA.Swan MK, Legris V, Tanner A, Reaper PM, Vial S, Bordas R, Pollard JR, Charlton PA, Golec JM, Bertrand JA. Acta Crystallogr D Biol Crystallogr. 2014 May; 70(Pt 5):1465-75. Epub 2014 Apr 30.
- Review [Bloom syndrome].[Nihon Rinsho. 2000]Review [Bloom syndrome].Kondo N. Nihon Rinsho. 2000 Jul; 58(7):1460-6.
- Review Bloom's Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition.[Mol Syndromol. 2017]Review Bloom's Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition.Cunniff C, Bassetti JA, Ellis NA. Mol Syndromol. 2017 Jan; 8(1):4-23. Epub 2016 Nov 5.
- Discovery of ML216, a Small Molecule Inhibitor of Bloom (BLM) Helicase - Probe R...Discovery of ML216, a Small Molecule Inhibitor of Bloom (BLM) Helicase - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...