

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

Although the induction of apoptosis can be an effective strategy for anti-cancer chemotherapy, emergence of drug resistance to existing pro-apoptotic drugs is a serious concern. Understanding the molecular mechanisms involved in acquiring drug resistance is of great interest for driving the discovery of new agents designed to thwart the escape routes tumor cells use to circumvent current therapy. The oncoproteins of the Bcl-2 family are activated in many forms of cancer, including the well-characterized lymphoid tumors multiple myeloma (MM) and chronic lymphoblastic leukemia (CLL). Bcl-2 family members regulate apoptosis induced by many cytotoxic compounds through multiple protein-protein interactions. The pro-survival protein Mcl-1 interacts with Bcl-2 family oncoproteins, including Bim, to oppose cell death. Mcl-1 is highly expressed in hematopoietic stem cells and is regulated by growth factors. Blocking the effects of Mcl-1, particularly through the inhibition of its interactions with pro-apoptotic oncogenes of the Bcl-2 family, is a promising approach for dramatically slowing tumor initiation, progression, and apoptosis resistance, especially in MM, where Mcl-1 is essential for tumor survival. We have used ultra-high throughput screening (uHTS) coupled with hit optimization strategy to discover potent and selective inhibitors of the Mcl-1/Bim interaction, including a designated probe compound, ML311. This new small molecule (also known as EU-5346) will be a useful tool for studying lymphoid tumorigenesis and to demonstrate the potential for using this strategy in therapies intended to bypass apoptosis resistance pathways that are activated in drug-resistant tumors.
Assigned Assay Grant #: 1 R43 CA135915-01 Fast Track
Screening Center Name & PI: The Scripps Research Institute Molecular Screening Center (SRIMSC), H Rosen
Chemistry Center Name & PI: SRIMSC, H Rosen
Assay Submitter & Institution: Michael Cardone, Eutropics Pharmaceuticals
PubChem Summary Bioassay Identifier: AID 2090
Probe Structure & Characteristics

| CID ML# SIDs | Target Name | IC50 [SID, AID] | Anti-Target | IC50 [SID, AID] | Fold Selective | Additional Assay(s) Name: [SID, AID] |
|---|---|---|---|---|---|---|
| CID 3384730 ML311 SID 131465452, SID 135383441, SID 49670212 (MLSMR) | Mcl1 (Biochem) | 0.31 μM [SID 135383441
AID 624393] 40%INH [SID 49670212, AID 2057] | Bcl-xL (Biochem) | >40 μM [SID 135383441, AID 624411] 16.7%INH [SID 49670212, AID 2129] | >129 | EC50values (μM) for SID 135383441: Mcl-1-1780 (cell-based target) 0.3 μM (AID 624396) Bcl-2-1863 1.1 μM (AID 624396) DHL-6 (cell-based target) 3.3 μM (AID 624396) NCI-H929 (cell-based target) 1.6 μM (AID 624396) DHL-10 (cell-based anti-target) 25 μM (AID 624396) % Apoptotic (DAPI staining) Mcl-1-1780:99% (0.5 μM) (AID 624396) Bcl-2-1863:21% (0.5 μM) (AID 624396) |
- †
Fold-selectivity was calculated as IC50 for anti-target/IC50 for the target, both biochemical assays
1. Recommendations for Scientific Use of the Probe
A small molecule inhibitor of the Mcl-1/Bim protein-protein interaction may halt growth of existing tumors that are resistant to pro-apoptotic drugs. It may also target tumors highly expressing Mcl-1. Researchers will use the probe in known models resistant tumors in cell cultures, including 3D Matrigel studies of tumor growth and in vivo studies using transgenic mice. The probe may potentiate the effects of other chemotherapeutic agents, delaying onset of resistance. The research community at large will use the probe to study how resistance mechanisms in lymphoid cancers may be circumvented.
2. Materials and Methods
All chemical reagents and solvents were acquired from commercial vendors. Reactions were monitored by LC/MS using two instruments, a Thermo/Finnegan LCQ Duo system with MS/MS capability and an Agilent 6140 quad system with 1200 LC. Analytical HPLC was used for quantitative purity assessment (Agilent 1200). Teledyne-Isco “combiflash” automated silica gel MPLC instruments were used for chromatographic purifications. A 400 Brüker MHz NMR instrument was used for NMR analysis. All assay protocols are reported in the relevant PubChem AIDs, provided in Table 1. Solubility, stability, and glutathione reactivity analyses were conducted in accordance with NIH guidelines. CYP450 inhibition and microsome stability analyses were performed as previously described17.
Table 1
All PubChem AIDs for the Mcl-1inhibitor project.
2.1. Assays
Descriptions of the assays follow Table 1.
Primary Assay
Mcl-1 Primary Assay
(see AID 2057, AID 2168, AID 2217, AID 2791, AID 493086, and AID 624393)
The purpose of the primary uHTS assay is to identify compounds that act as Mcl-1/Bim inhibitors. This biochemical assay monitors binding of the BH3-only protein Bim to the BH3 binding pocket of human Mcl-1. In this fluorescence polarization (FP)-based assay, GST/Mcl-1 fusion protein is incubated with FITC-BH3-Bim peptides in the presence of test compounds. Binding of peptide to Mcl-1 target protein increases the effective molecular mass of the peptide, slowing its rotation and increasing millipolarization (mP) units in the well. As designed, compounds that bind Mcl-1 and prevent its binding with Bim peptide increase the proportion of free to bound peptides, thereby reducing mP in the well. Compounds are tested in singlicate at a final nominal concentration of 10.9 μM. The short name for the primary assay is: MCL-1 BIM_INH_FP_1536_1X%INH; the descriptive name is: Fluorescence polarization-based primary biochemical high throughput screening assay to identify inhibitors of myeloid cell leukemia sequence 1 (MCL-1) interactions with BIM-BH3 peptide. Assay IDs in Pubchem are as follows: AID 2057 (PRUN), AID 2168 (CRUN), AID 2217 (DRUN), AID 2791 (Powder IC50 run by SRIMSC); AID 493086, AID 624393 (powders, run by assay provider).
Secondary Assays
Bcl-xL Assay (HTS Counterscreen, Biochemical Assay)
(see AID 2129, AID 2166, AID 2218, AID 2790, and AID 624411)
The purpose of the counterscreen was to identify compounds that act as Bcl-xL inhibitors. This fluorescence polarization (FP)-based assay monitors binding of the BH3-only protein Bim to the BH3 binding pocket of Bcl-xL. GST-Bcl-xL fusion protein is incubated with FITC-BH3-Bim peptides, in the presence of test compounds. Binding of peptide to Bcl-xL target protein increases the effective molecular mass of the peptide, slowing its rotation and increasing millipolarization (mP) units in the well. As designed, compounds that inhibit Bcl-xL will prevent binding of peptide to Bcl-xL protein, and increase the ratio of free to bound peptides, thereby reducing mP in the well. Compounds are tested in singlicate at a final nominal concentration of 10.9 μM. The short name for this assay is: BCLXLBIM_INH_FP_1536_1X%INH; the descriptive name is: Biochemical high throughput screening assay to identify inhibitors of Bcl-2-related protein, long isoform (Bcl-xL). Assay IDs in Pubchem are as follows: AID 2129 (Full deck), AID 2166 (3X CSRUN), AID 2218 (DCSRUN), AID 2790 (powder IC50), and 624411 (powders, run by assay provider).
Other Secondary (SAR) Assays (Cellular Assays)
(see AID 602164, AID 493058, AID 493062 and AID 493059)
A number of secondary assays were used in this probe development effort. Secondary assay AID 602164 was utilized to determine the cell-killing abilities of probe candidates in cells which are either highly Mcl-1 primed or highly Bcl-2 primed, and therefore susceptible to apoptosis induction by Mcl-1 and Bcl-2 inhibitors. The short name for this assay is: MCL1-APOP_ACT_ABS_96_3XEC50 MCL1-BCL2 cells; the descriptive name is: MCL-1 inhibitor specificity and apoptosis pathway induction: Cell-based assay to identify compounds that are preferentially active in causing apoptosis in Mcl-1 primed vs. Bcl-2 cell lines. The purpose of this assay is to identify compounds that are preferentially active in cells that are Mcl-1 dependent (Mcl-1-1760) compared to Bcl-2 dependent (Bcl-2-1863).
Another secondary assay, the cytotoxicity assay AID 493058, was utilized to identify probe candidates that exert their apoptotic effects in cell lines by a Bcl-2 pathway dependent mechanism (including Mcl-1, Bcl-2, or Bcl-xL). The Bax/Bak-functional cell lines NCI-H929 and DHL-6 and the Bax/Bak deficient cell line DHL-10 were used. Mcl-1-selective compounds should have minimal effects in DHL-10 cells, as Bax and Bak are found downstream of Mcl-1 and are essential components of the intrinsic apoptosis pathway. The short name for this assay is: DHL10-CYTOX_ACT_FLINT_96_3XEC50 Round 0 Bax/Bak-deficient; the descriptive name is: Late stage assay provider results from the probe development effort to identify Mcl-1/Bim inhibitors: absorbance-based cell-based assay to identify compounds that are preferentially active in causing apoptosis in MCL-1 primed vs. Bax/Bak deficient DHL-10 cells.
Another secondary assay supporting probe development is AID 493062. The descriptive name for this assay is, “Late stage assay provider results from the probe development effort to identify Mcl-1/Bim inhibitors: fluorescence-based cell-based assay to identify compounds that preferentially activate caspase in 2B4/MCL-1 vs. 2B4/BCL-2 cells”. The purpose of this assay is to identify compounds that induce caspase activation in a Mcl-1 dependent fashion and that exhibit selectivity for Mcl-1 over Bcl-2 as reflected by enhanced potency in Mcl-1 dependent cell line compared to a Bcl-2 dependent cell line. The Bcl-2 specific compound ABT-737 will be used as a control for Bcl-2 specific compounds, since ABT-737 does not bind to Mcl-1. Another secondary assay supporting probe development is the DAPI Apoptosis Assay (AID 493059 and AID 624396). The short name for this assay is: APOPTOSIS_ACT_DAPI_FLUOR_3X%ACT MCSRUN Round 0; the descriptive name is: Late stage assay provider results from the probe development effort to identify Mcl-1/Bim inhibitors: fluorescence-based cell-based cytotoxicity assay using 4′,6-diamidino-2-phenylindole (DAPI) to identify compounds that are pro-apoptotic in cancer cells grown in culture. The purpose of this assay is to identify compounds that activate caspase 3 and 7 resulting in apoptotic nuclear morphology. As activators of apoptosis, the probes should increase caspase activity due to loss of mitochondrial membrane potential and cytochrome c release. As this assay is highly time consuming and very low throughput, it was performed only on the final probe candidate.
AID 602164; Assay short name: MCL1-APOP_ACT_ABS_96_3XEC50 MCL1-BCL2 cells
Descriptive Name: MCL-1 inhibitor specificity and apoptosis pathway induction: Cell-based assay to identify compounds that are preferentially active in causing apoptosis in Mcl-1 primed vs. Bcl-2 cell lines.
Protocol: The mouse leukemia-derived cell lines MCL-1-1780 and BCL-2-1863 (Ryan et al., Proc. Nat. Acad. Sci USA, 107, 12895–12900) were obtained from Anthony Letai of the Dana Farber Cancer Institute, Boston, MA. Cells were grown in RPMI 1640 medium (GIBCO-BRL) with 2 mM L-glutamine, 4.5 g/L glucose, 1.0 mM sodium pyruvate and 5% fetal bovine serum. Cells were expanded in tissue culture in appropriate media and then sub-cultured into 96-well plates at a seeding density of 20,000 cells per well. After incubation for 24 hours, cells were treated with compounds that are titrated into appropriate medium with FCS. Seven concentrations of compound were used; the standard concentration points used were: 25 μM, 12.5 μM, 6.24 μM, 3.1 μM, 1.6 μM, 0.8 μM, and 0.4 μM. A second round of assays were performed with active inhibitors, in which compound concentrations of: 5.0 μM, 2.5 μM, 1.25 μM, 625 nM, 313 nM, 156 nM, and 78 nM were used. All assays were run in triplicate for each compound at each concentration and averages were calculated. Cells were treated for 48 hours and scored for viability using the MTS assay (Promega). Growth inhibition was calculated as a percentage of control cell growth. Growth was determined by measuring the A570 (control cells) - A570 (treated cells)/A570 (control cells). GI50 values were calculated using Graphpad Prism software.
AID 493058; Assay short name: MCL1-APOP_ACT_ABS_96_3XEC50 Bax/Bak-def cells
Descriptive Name: MCL-1 inhibitor specificity and apoptosis pathway induction: Cell-based assay to identify compounds that are preferentially active in causing apoptosis in Bcl-2/Mcl-1 primed cells (DHL-6 or NCI-929) vs. Bax/Bak deficient cells (DHL-10).
Protocol: The lymphoid-derived cell lines DHL-6 (Bax/Bak functional, on-target indicator) and DHL-10 (Bax/Bak deficient, anti-target indicator) were obtained from Anthony Letai of the Dana Farber Cancer Research Institute, Boston, MA20. The myeloid derived cell line NCI-H929 (Bax/Bak functional, on-target indicator) was obtained from the NIH/NCI cell repository. Cells were grown in RPMI 1640 medium (GIBCO-BRL) with 2 mM L-glutamine, 4.5 g/L glucose, 1.0 mM sodium pyruvate and 5% fetal bovine serum. Cells were expanded in tissue culture in appropriate media and then sub-cultured into 96-well plates at a seeding density of 20,000 cells per well. After incubation for 24 hours, cells were treated with compounds that are titrated into appropriate medium with FCS. Seven concentrations of compound were used; the standard concentration points used were: 25 μM, 12.5 μM, 6.24 μM, 3.1 μM, 1.6 μM, 0.8 μM, and 0.4 μM. A second round of assays were performed with active inhibitors, in which compound concentrations of: 5.0 μM, 2.5 μM, 1.25 μM, 625 nM, 313 nM, 156 nM, and 78 nM were used. All assays were run in triplicate for each compound at each concentration and averages were calculated. Cells were treated for 48 hours and scored for viability using the MTS assay (Promega). Growth inhibition was calculated as a percentage of control cell growth. Growth was determined by measuring the A570 (control cells) - A570 (treated cells)/A570 (control cells). GI50 values were calculated using Graphpad Prism software.
AID 493062; Assay short name: CASPASE_ACT_FLUOR_96_3XEC50
Descriptive Name: Counterscreen for MCL-1 selective inhibitors: Whole-cell caspase activation assay in 2B4/Mcl-1 and 2B4/Bcl-2 to determine Bcl-2 family target selectivity and induction of downstream activators of apoptosis.
See also assay short name: CASPASE-2B4_ACT_FLUOR_96_3XEC50 MDCSRUN Round 0; Descriptive name: Late stage assay provider results from the probe development effort to identify Mcl-1/BIM inhibitors: fluorescence-based cell-based assay to identify compounds that preferentially activate caspase in 2B4/MCL-1 vs. 2B4/BCL-2 cells.
Protocol: As activators of apoptosis, the probes should increase caspase activity due to loss of mitochondrial membrane potential and cytochrome c release. We conducted caspase assays in the paired cell lines 2B4/Mcl-1 and 2B4/Bcl-2 to show that caspase activation is Mcl-1-specific. Cells were incubated at a fixed time interval and caspase activation is assessed with a lumogenic protease assay (Promega Caspase-Glo 3/7). A 3-fold or higher shift in the EC50 for caspase activation is preferred. To visualize the percentage of cells in which there is caspase activity treated cells were fixed with methanol and stained with the DNA binding DAPI (4′,6-diamidino-2-phenylindole) and observed using a fluorescence microscope. The DNA bound dye emits at 456 nm when excited by 350 nm light. Caspase-mediated cleavage of nucleosomal proteins causes condensed, pycnotic nuclear morphology that is easily scored. Percentages of apoptotic cells so scored were compared to positive control samples. Staurosporine was used as positive control. In addition, the fluorescent cationic dye JC-1 was be used to measure the efficacy of the drug on mitochondrial integrity. The JC-1 assay will be conducted in 2B4 cell lines to detect MOMP. A 3-fold or higher shift in the EC50 for mitochondrial outer-membrane permeabilization in 2B4/Mcl-1 vs. 2B4/Bcl-2 mitochondria is preferred.
AID 493059; Assay short name: APOPTOSIS_ACT_DAPI_FLUOR_EC50
Descriptive Name: Late stage assay provider results from the probe development effort to identify Mcl-1/BIM inhibitors: fluorescence-based cell-based cytotoxicity assay using 4′,6-diamidino-2-phenylindole (DAPI) to identify compounds that are pro-apoptotic in cancer cells grown in culture.
Protocol: The mouse leukemia-derived cell lines Mcl-1-1780 and Bcl-2-1863 (Ryan et al., Proc. Nat. Acad. Sci USA, 107, 12895–12900) were obtained under license from Dana Farber Cancer Institute. Cells were grown in RPMI 1640 medium (GIBCO-BRL) with 2 mM L-glutamine, 4.5 g/L glucose, 1.0 mM sodium pyruvate and 5% fetal bovine serum. Cells were expanded in tissue culture in appropriate media and then sub-cultured into 96-well plates at a seeding density of 20,000 cells per well. After incubation for 24 hours, cells were treated with compounds that are titrated into appropriate medium with FCS. Four concentrations of compound were used: 1 μM, 500 nM, 250 nM, and 125 nM. Assay was run in triplicate for each compound at each concentration and averages were calculated. Cells were treated with compound for 24 hours, then fixed with methanol and stained with the DNA-binding agent DAPI (4′,6-diamidino-2-phenylindole). The cells were then examined by fluorescent microscopy; cells in which caspase 3 and caspase 7 have not been activated will emit at 456 nM when excited by 350 nM light. Caspase-mediated cleavage of nucleosomal proteins causes condensed, pycnotic nuclear morphology that may be measured by cell counting. The percentage of cells which exhibit apoptosis was calculated and reported.
2.2. Probe Chemical Characterization
The chemical structure of the probe was verified by analysis of its 400 MHz 1H NMR spectra (Figure 1) obtained on a Brüker 400 MHz instrument. The chemical structure was also corroborated by its LC/MS molecular ion (calc for M+1: 416.2, found 416.2, using an Agilent 1200/6140 multimode quadrupole rapid resolve system) as well as its expected fragmentation patterns. Purity was measured at >95% (LC/MS analysis, confirmed by analytical HLPC analysis; HPLC purity data is shown in Figure 2). HPLC data was obtained using an Agilent 1200 analytical HPLC with an Agilent Eclipse XDB-C18 column, 4.6×150mm. The HPLC solvents used were acetonitrile and water with 0.1% formic acid added to each mobile phase as the pH modifier.

Figure 1
NMR Spectra ML311. Full spectrum (top) and aryl region expansion (bottom).
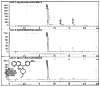
Figure 2
Analytical RP HPLC Spectra for ML311. At 254 nM, 280nM, and 320 nM, purity >95%.
The solubility of the probe in PBS at pH 7.4 was determined to be >2.7 μM. Solubility was higher (~20 μM) in conditions more reflective of the cell-based assay environment (DMEM, 10% FBS). Importantly, its solubility is fully adequate to provide the high potency seem in multiple cell-based assays (0.4–5 μM) and is also adequate for broad use as a biological probe to be used in a variety of aqueous-based media.
The probe has a half-life of 10 hours in PBS at room temperature when tested at 10 μM. Disappearance of the LC peak for the ML311 is not altered by addition of excess glutathione, indicating that ML311 is not thiol-reactive under physiologically-relevant conditions.
ML311 is stable in DMSO solution at room temperature (no erosion of peak intensity after 7 days) and is also stable as a free base dry powder.
NMR and HLPC spectra are shown in Figures 1 and 2.
The compounds listed in Table 2 have been submitted.
Table 2
Probe and submitted analogs.
2.3. Probe Preparation
General Synthesis Scheme. The probe compound and all analogs were prepared efficiently in a one step/one pot procedure from commercially available starting materials, as shown in the general scheme in Figure 3. In this procedure, termed the Betti reaction18 of 8-hydroxyquinolines19, an aldehyde A and amine B condense to form an iminium ion intermediate C, which then reacts with the 8-hydroxyquinoline D at the 7-position to give the product E. The probe analogs and SAR compounds were prepared by this procedure using available structural variants of reagents A, B, and D.

Figure 3
Betti reaction of 8-hydroxyquinolines (X = N): general synthesis scheme for preparing the probe ML311 and all analogs.
Detailed experimental procedures for the preparation of 7-((4-Ethylpiperazin-1-yl)(4-(trifluoromethyl)phenyl)methyl)quinolin-8-ol, or ML311. (See Figure 4.) The general synthesis scheme from Figure 3 was followed.

Figure 4
Detailed experimental procedures for the preparation of ML311.
A mixture of quinolin-8-ol (470 mg, 3.24 mmol), 4-(trifluoromethyl)benzaldehyde (564 mg, 3.24 mmol) and 1-ethylpiperazine (0.41 mL, 3.24 mmol) was heated overnight at 110 ˚C in an oil bath. The solution was cooled to room temperature and the mixture was precipitated in ethanol. The solid was collected by filtration to yield the title compound (471 mg, 35% yield) in high purity (>95%) as judged by 1H NMR and LCMS analysis. The 1H NMR spectrum and analytical HLPC trace were shown in section 2.2. Supporting data: MS (M+1) cal’d = 416.2, found 416.2. 1H NMR (400MHz, DMSO-d6) δ 10.17 (br s, 1H), 8.82 (dd, J = 1.6, 4.4 Hz, 1H), 8.25 (dd, J = 1.6, 8.4 Hz, 1H), 7.71–7.63 (m, 5H), 7.51 (dd, J = 4.2, 8.2 Hz, 1H), 7.39 (d, J = 8.8 Hz, 1H), 5.05 (s, 1H), 2.49–2.29 (vbr s, 8H), 2.29 (q, J = 7.2 Hz, 2H), 0.95 (t, J = 7.2 Hz, 3H).
3. Results
3.1. Dose Response Curves for Probe
ML311 has robust on-target activity in biochemical assays (see Figure 5 for target and anti-target curves). The primary criteria of the probe report have been achieved: ML311 displays >100× selectivity in a biochemical assay (fluorescence polarization) for Mcl-1 over Bcl-xL. In addition, as shown in Figure 6, ML311 induces cell death in a highly active Mcl-1 cell line (Mcl-1/1780, EC50 = 0.3 μM). The compound does not induce appreciable cell death in the anti-target cell based assay (DHL-10) even at the highest dose tested (5 μM), and in other runs for a previous batch (SID 131465452) this lack of anti-target effect was verified at higher concentrations (to 25 μM). There is thus at least 8-fold selectivity for the DHL-6 cell line versus the DHL-10 cell line, meeting the criteria established for probe in the CPDP of greater than 3-fold selectivity. ML311 is also approximately 4-fold more active in Mcl-1-1780 cells than vs. the highly Bcl-2 active cell line Bcl-2-1863 (EC50 =1.1 μM), meeting another probe development objective. Batch-to-batch consistency in activity is seen in all assay formats, as indicated in Figures 5 and 6.

Figure 5
Primary target, biochemical assay (Mcl-1). Anti-target, biochemical counterscreen (Bcl-xL)

Figure 6
Cell-based assays. Targets and DHL-10 anti-target
3.2. Cellular Activity
The probe potently halts viability of several types of Mcl-1 primed cells, including MCL-1-1780 (EC50 = 0.31 μM), DHL-6 (EC50 = 3.3 μM), and NCI-H929 (EC50 = 1.6 μM), with generally high maximal effect (>80%). All of the cell-based curves were shown previously in Section 3.1 (Figure 6).
ML311 also displayed activity in a leukemia-derived cell line particularly reliant upon Bcl-2 function (Bcl2-1863, EC50 = 1.1 μM). Targeting of Bcl-2 is considered highly relevant for the treatment of a number of hematological malignancies, as evidenced by the successes observed with ABT-263 (Navitoclax) and related compounds. However, for the purposes of this probe development effort, some selectivity over Bcl-2 was desired, with a selectivity goal of 3X for Mcl-1 1780 versus Bcl-2 1863 cell lines. The exclusion of all Bcl-2 activity was not sought, merely the objective as set forth in the CPDP was to obtain compounds with Mcl-1 dependant effects and with a bias for Mcl-1 over Bcl-1, a profile unknown for a small molecule at the time when this effort was initiated. This goal was achieved with the development of ML311.
The finding that the DHL-10 anti-target is largely unaffected is highly significant (EC50 >25 μM, Figure 6). DHL-10 is Bax/Bak deficient and is properly unaffected, since it is incapable of the Mcl-1/Bim interaction. This result shows the probe’s promise for useful on-target specificity and selective cytotoxicity in the mode of action.
To assess cell-based antitumor activity more broadly, ML311 has been submitted for analysis in the the NCI’s CTD2 (Cancer Target Discovery and Development program) through the Broad Institute and in the NCI60 panel. The CTD2 program involves screening 1,000 well-characterized cancer cell lines with results made publicly available.
The NCI60 screening results were completed and are summarized in Figure 7. ML311 has strong growth inhibitory effects in many cell lines, with GI50 <900 nM for nine cell types, and <2 μM for 14 additional types (not shown). Mcl-1 is known to be highly expressed in many forms of lung, prostate, breast, ovarian, renal cancers, and gliomas by mRNA analysis.31 Correlation of NCI60 activity shown in Figure 7 with intrinsic Mcl-1 mRNA levels is unclear, though mRNA levels are also not necessarily reflective of protein levels as determined by other means, such as Western blot analysis. Also certain tumor types with only modest Mcl-1 levels may be strongly affected by dampening the activity of Mcl-1 present.

Figure 7
NCI60 growth inhibition.
3.3. Profiling Assays
The cell-based assays used in screening and in support of our lead optimization efforts define a clear mode of action for ML311: disruption of the interaction of Mcl-1 with Bim in the BH3 binding region of Mcl-1. To support the selectivity for this mode of action hypothesis, we studied the properties of the probe and its precursors.
Analysis of PubChem assay results for the initial HTS hit, CID 2966040, suggests that this compound (and perhaps the scaffold itself) is not assay-promiscuous, with activity in 20 of 508 Pubchem assays (3.9%), when Mcl-1-related assays are excluded from the analysis. The lack of broad activity for this HTS hit was one factor leading to the selection of this scaffold for probe development.
The lack of activity of CID 2966040 and many of its analogs in the Bcl-xL counterscreen and the DHL-10 cell-based antitarget counterscreen also provides further evidence of target-specific activity. Analysis of the structure using online cheminformatics tools to assess shape similarity to known ligands (molinspiration bioactivity analysis) 31 suggests a low probability that ML311 is a significantly potent GPCR ligand, ion channel modulator, kinase inhibitor, nuclear receptor ligand, protease inhibitor, or other enzyme inhibitor.
To gauge selectivity in a broader sense, ML311 was submitted for analysis in a lead profiling screen with Ricerca, a screen profiling binding to 67 protein targets of therapeutic and/or toxicological interest (Figure 8). The probe is inactive vs. most targets. Moderate activity (50–80% at 10 μM) was noted for certain GPCRs and ion channels, as indicated. For a few targets, notably hERG, significant effects are seen. This potential for GPCR and ion-channel mediated effects should be considered when using ML311 in animal studies. Reduction of these off-target effects is desirable in follow-up compounds.

Figure 8
Lead profiling screen results summary.
4. Discussion
4.1. Comparison to Existing Art and How the New Probe is an Improvement
At the onset of the uHTS campaign no published prior art small molecules were known to selectively disrupt the interaction of Mcl-1 and Bim. The relevant prior art consisted of three non-selective compounds (Table 3).
Table 3
Comparison of EU-517, obatoclax, gossypol, and ML311.
The aryl sulfone termed “EU-517” and analogs were discovered at Harvard in a screen for inhibitors of the interaction of BH3 with Bcl-xL33. This compound has modest potency vs. both Bcl-xL and Mcl-1-primed cell lines (11.2, 5.3 μM, respectively). The assay provider (Eutropics) has evaluated the activity of EU-517 against Mcl-1 in in vitro protein binding and cell-based assays. This compound is effective in killing tumor cells in vitro with EC50 values in the micromolar range33. In unpublished work, the assay provider has found that EU-517 aids survival of mice bearing lymphatic tumors and tumors of the spleen. The specific mechanism of action of this series of compounds, including EU-517, was validated by NMR in the lab of Dr. Gerhard Wagner34, who demonstrated binding to the same hydrophobic groove as the BH3 peptides. Further, NMR chemical shift perturbation studies were used to map the interaction interfaces and to define the conformational space for molecular modeling.
The second relevant prior art compound to consider is obatoclax, which has progressed into phase II trials. It is reported to cause neurological symptoms (euphoria) in CLL patients and neural toxicity in mice35–37, in vivo toxicities inconsistent with its action as a BH3 mimetic. The conjugated pyrrole structure is also related to ATP-competitive kinase inhibitors, which could explain some off-target activity, including added cytotoxicity. The absence of thrombocytopenia, demonstrated to be an on-target effect of Bcl-xL inhibition, in two independent studies is also telling. We obtained a sample of obatoclax and found that it has ~5-fold lower activity in the Mcl-1 FP assay than does ML311 (1.5 μM for obatoclax, 0.31 μM for ML311).
A third prior art compound to consider is gossypol (AT-101), which like obatoclax exhibits moderate pan-Bcl-2 inhibitory activity, blocking Mcl-1 action along with that of its structural homologs Bcl-2 and Bcl-xL38. Recent characterization of the mechanism of action for gossypol and obatoclax, as well as findings of significant yet poorly understood off-target cytotoxicity, raise concerns with respect to their potential for clinical success. Despite their pan-Bcl-2 activity, these compounds (unlike ML311) kill cells in a Bax-Bak independent fashion and do not induce caspase-9 dependent cell death39. Instead, they likely function through a non-apoptotic pathway as mitochondrial toxins, raising a concern of causing non-mechanism-based toxicities in vivo. Indeed, gossypol is used as a male contraceptive in China.
For all of these compounds the lack of a clear, single, and selective mechanism of action diminishes the feasibility of identifying patient populations likely to benefit from treatment.
There have been a number of commercial efforts to develop drugs that inhibit BH3 binding to anti-apoptotic proteins, without selectivity for Mcl-1. Most notable is a program at Abbott that has yielded a very active Bcl-2/Bcl-xL inhibitor ABT-263 now in clinical trials13. This compound however, has been shown to be ineffective in cancer cells that express abundant Mcl-140,41. This is a significant problem given the key role that Mcl-1 plays in blood cancer onset and in cellular chemoresistance42–44. Elevated expression of Mcl-1 and elevated Mcl-1/Bax ratio correlates with chemoresistance in CLL and NHL patients, pointing to the importance of targeting Mcl-1 as a therapeutic strategy. There are serious obstacles to the successful development of ABT-263, including that it induces thrombocytopenia in patients due to inhibition of Bcl-xL, which is essential for platelet survival45,46. This on-target toxicity requires precisely calibrated dosing regimens to gain a tolerable therapeutic window.
Given the high interest in targeting Bcl-2 family proteins, it is not surprising that additional reports have surfaced since the onset of our probe development efforts. In July 2010 a structure related to ML311 termed “compound 65” was disclosed47. No biochemical data or single agent cellular data was reported for compound 65, rather only a positive synergy with the kinesin spindle protein inhibitor ARRY-520 was disclosed. We independently studied this compound (CID 3625042, SID 125001187, Figure 9) in the course of our probe development efforts and found it to be slightly less potent in all target assays than is ML311 (compound 65 has IC50 = 1.1 μM for Mcl-1 in the FP assay, 3.4, 6.0, and 4.2 μM for Mcl-1-1780, DHL-6, and NCI-H929 cells, respectively; compare with values for ML311 of 0.31 μM in the FP assay and 0.3, 3.3, and 1.6 μM in the cell-based assays).
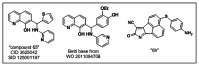
Figure 9
Recently-published Mcl-1 inhibitors. (Reported after our uHTS.)
In 2011, a group from the Dana-Farber Cancer Institute published a patent application claiming uses of twelve scaffolds with activity vs. Mcl-1, as based on single-point screening data. One of the twelve described scaffolds is the 8-hydoxyquinolines48. The same group has also published data on non small molecule “stapled peptides” as Mcl-1 inhibitors49. These compounds are not freely available for use, and no data is given to determine their usefulness as probes, such as IC50/EC50 values for biochemical and cell-based activity, cell-based selectivity, etc. A representative of the seven compounds in the 8-hydroxyquinoline class is also shown in Figure 9. The cursory and incomplete description of the activity of the compounds, as well as the lack of discussion as to whether any of them may be preferred over the numerous other compounds in the eleven other scaffolds that are exemplified in that patent application, fails to establish the compounds in the Dana-Farber disclosure as prior art that would seriously limit the usefulness of ML311. In addition, this data was published after the initiation of the current MLPCN effort and the identification of the hit scaffold.
Also, in 2011 an academic research team in China reported efforts to target Mcl-1 by modification of a scaffold previously shown to be a dual inhibitor of Bcl-2 and Mcl-150. The most potent compound in the series, compound 6h (Figure 9) showed low nanomolar competitive binding affinity with Mcl-1 in an ELISA assay, ~60-fold selectivity vs. Bcl-2, and impressive cytotoxicity vs. three tumor cell lines by an MTT assay (IC50 = 12–18 nM). It is not clear, however, if the cytotoxicity is Mcl-1-dependent in the absence of more control studies, particularly since related compounds with lesser affinity for Mcl-1 were also cytotoxic to a similar extent. A serious issue is the poor drug-likeness and multiple structure alerts of this large, mostly planar, and highly hydrophobic structure, which is similar to other large heteroaromatic compounds that act as cytotoxins through DNA intercalation.
5. References
- 1.
- McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Update. 2008;11(4–5):164–79. [PubMed: 18818117]
- 2.
- Reed JC. Drug insight: cancer therapy strategies based on restoration of endogenous cell death mechanisms. Nat Clin Pract Oncol. 2006;3(7):388–98. [PubMed: 16826219]
- 3.
- Cory S, Adams JM. Killing cancer cells by flipping the Bcl-2/Bax switch. Cancer Cell. 2005;8(1):5–6. [PubMed: 16023593]
- 4.
- Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644(2–3):83–94. [PubMed: 14996493]
- 5.
- Redzepovic J, Weinmann G, Ott I, Gust R. Current trends in multiple myeloma management. J Int Med Res. 2008;36(3):371–86. [PubMed: 18534118]
- 6.
- Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL, Bataille R, Amiot M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. 2002;100(1):194–9. [PubMed: 12070027]
- 7.
- Hussein S-R, Cheney C, Johnson A, Lin T, Grever M, Caliguiri M, Lucas D, Byrd J. Mcl-1 is a Relevant Therapeutic Target in Acute and Chronic Lymphoid Malignancies: Down-Regulation Enhances Rituximab-Mediated Apoptosis and Complement-Dependent Cytotoxicity. Clin Cancer Res. 2007;13:2144–2150. [PubMed: 17404098]
- 8.
- Inoue S, Walewska R, Dyer MJ, Cohen GM. Downregulation of Mcl-1 potentiates HCACi-mediated apoptosis in leukemic cells. Leukemia. 2008;22(4):819–825. [PubMed: 18239621]
- 9.
- Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci USA. 1993;90(8):3516–20. [PMC free article: PMC46331] [PubMed: 7682708]
- 10.
- Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307(5712):1101–4. [PubMed: 15718471]
- 11.
- Brunelle JK, Ryan J, Yecies D, Opferman JT, Letai A. MCL-1–dependent leukemia cells are more sensitive to chemotherapy than BCL-2–dependent counterparts. J Chem Biol. 2009;187(3):429. [PMC free article: PMC2779245] [PubMed: 19948485]
- 12.
- Letai A. Pharmacological manipulation of Bcl-2 family members to control cell death. J Clin Invest. 2005;115(10):2648–55. [PMC free article: PMC1236690] [PubMed: 16200198]
- 13.
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–81. [PubMed: 15902208]
- 14.
- Vogler M, et al. Different forms of cell death induced by putative Bcl2 inhibitors. Cell Death and Differentiation. 16:1030–1039. [PubMed: 19390557]
- 15.
- Verdine GL, Walensky LD. The challenge of drugging undruggable targets in cancer: lessons learned from targeting Bcl-2 family members. Clin. Cancer Res. 13:7264–7270. [PubMed: 18094406]
- 16.
- Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Aleo MD. Structural Alert/Reactive Metabolite Concept as Applied in Medicinal Chemistry to Mitigate the Risk of Idiosyncratic Drug Toxicity: A Perspective Based on the Critical Examination of Trends in the Top 200 Drugs Marketed in the United States. Chem ResToxicol. 2011;24:1345–1410. [PubMed: 21702456]
- 17.
- Brothers SP, Saldanha SA, Spicer TP, Cameron M, Mercer BA, Chase P, McDonald P, Wahlestedt C, Hodder PS. Selective and brain penetrant neuropeptide Y Y2 receptor antagonists discovered by whole-cell high-throughput screening. Mol Pharmacol. 2010;77(1):46–57. [PMC free article: PMC2802430] [PubMed: 19837904]
- 18.
- Betti M. Org Synth Coll Vol I. 1941;1:381.
- 19.
- Phillips JD. The reactions of 8-quinolinol. Chem. Rev. 1956;56:271–297.
- 20.
- Certo M, Moore VDG, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9(5):351–65. [PubMed: 16697956]
- 21.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews. 2001;46:3–26. 11259830. [PubMed: 11259830]
- 22.
- Walters WP. Going further than Lipinski’s rule in drug design. Expert Opinion on Drug Discovery. 2012;7(2):99–107. 22468912. [PubMed: 22468912]
- 23.
- Rishton GM. Nonleadlikeness and leadlikeness in biochemical screening. Drug Discovery Today. 2003;8(2):86–96. 12565011. [PubMed: 12565011]
- 24.
- Benigni R, Bossa C. Mechanisms of Chemical Carcinogenicity and Mutagenicity: A Review with Implications for Predictive Toxicology. Chem Rev. 2011;111(4):2507–2536. 21265518. [PubMed: 21265518]
- 25.
- Cardellina JH, Vieira RC, Eccard V, Skerry J, Montgomery V, Campbell Y, Roxas-Duncan V, Leister W, LeClair CA, Maloney DJ, Padula D, Pescitelli G, Khavrutskii I, Hu X, Wallqvist A, Smith LA. Separation of Betti Reaction Product Enantiomers: Absolute Configuration and Inhibition of Botulinum Neurotoxin A. ACS Med Chem Lett. 2011;2:396–401. [PMC free article: PMC3217201] [PubMed: 22102940]
- 26.
- Cardellicchioa C, Capozzib MAM, Naso F. The Betti base. The awakening of a sleeping beauty. Tetrahedron: Asymmetry. 2010;21(5):507–517.
- 27.
- Betti M. Cleavage/fission of the naphtholbenzylamine into its optical antipodes. Gazz Chim Ital. 1906;36(II):392–394.
- 28.
- Cardellicchio C, Ciccarella G, Naso F, Schingaro E, Scordari F. The Betti base: absolute configuration and routes to a family of related chiral nonracemic bases. Tetrahedron: Asymmetry. 1998;9:3667–3675.
- 29.
- Dong Y, Li R, Lu J, Xu X, Wang X, Hu Y. An efficient kinetic resolution of racemic Betti base based on an enantioselective N,O-deketalization. J Org Chem. 2005;70:8617–8620. [PubMed: 16209623]
- 30.
- Guengerich FP. Cytochrome P450 and Chemical Toxicology. Chem Res Toxicol. 2008;21:70–83. [PubMed: 18052394]
- 31.
- Placzek WJ, Wei J, Kitada S, Zhai D, Reed JC, Pellecchia M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death and Disease. 2010:1–9. [PMC free article: PMC3032312] [PubMed: 21364647]
- 32.
- see http://www
.molinspiration.com/ for free online cheminformatics software. - 33.
- Verdine GL, Walensky LD. The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin Cancer Res. 2007;13(24):7264–70. [PubMed: 18094406]
- 34.
- Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan J. Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat Cell Biol. 2001;3(2):173–82. [PubMed: 11175750]
- 35.
- Lugovskoy AA, Degterev AI, Fahmy AF, Zhou P, Gross JD, Yuan J, Wagner G. A novel approach for characterizing protein ligand complexes: molecular basis for specificity of small-molecule Bcl-2 inhibitors. J Am Chem Soc. 2002;124(7):1234–40. [PubMed: 11841292]
- 36.
- O’Brien SM, Claxton DF, Crump M, Faderl S, Kipps T, Keating MJ, Viallet J, Cheson BD. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood. 2009;113(2):299–305. [PMC free article: PMC4968372] [PubMed: 18931344]
- 37.
- Trudel S, Li ZH, Rauw J, Tiedemann RE, Wen XY, Stewart AK. Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015-070) in multiple myeloma. Blood. 2007;109(12):5430–8. [PubMed: 17332241]
- 38.
- Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Estrov E, Bornmann W, Kantarjian H, Viallet J, Samudio I, Andreeff M. Mechanisms of Antileukemic Activity of the Novel Bcl-2 Homology Domain-3 Mimetic GX15-070 (Obatoclax). Cancer Research. 2008;68:3413–3420. [PMC free article: PMC4096127] [PubMed: 18451169]
- 39.
- Campas C, Cosialls AM, Barragan M, Iglesias-Serret D, Santidrian AF, Coll-Mulet L, de Frias M, Domingo A, Pons G, Gil J. Bcl-2 inhibitors induce apoptosis in chronic lymphocytic leukemia cells. Exp Hematol. 2006;34(12):1663–9. [PubMed: 17157163]
- 40.
- Vogler M, Weber K, Dinsdale D, Schmitz I, Schulze-Osthoff K, Dyer MJ, Cohen GM. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. 2009;16(7):1030–9. [PubMed: 19390557]
- 41.
- Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67(2):782–91. [PubMed: 17234790]
- 42.
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10(5):389–99. [PMC free article: PMC2953559] [PubMed: 17097561]
- 43.
- Cho-Vega JH, Rassidakis GZ, Admirand JH, Oyarzo M, Ramalingam P, Paraguya A, McDonnell TJ, Amin HM, Medeiros LJ. MCL-1 expression in B-cell non-Hodgkin’s lymphomas. Hum Pathol. 2004;35(9):1095–100. [PubMed: 15343511]
- 44.
- Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. 2002;99(6):1885–93. [PubMed: 11877256]
- 45.
- Michels J, O’Neill JW, Dallman CL, Mouzakiti A, Habens F, Brimmell M, Zhang KY, Craig RW, Marcusson EG, Johnson PW, Packham G. Mcl-1 is required for Akata6 B-lymphoma cell survival and is converted to a cell death molecule by efficient caspase-mediated cleavage. Oncogene. 2004;23(28):4818–27. [PubMed: 15122313]
- 46.
- Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, Iciek LA, Morgan SJ, Nasarre MC, Nelson R, Preusser LC, Reinhart GA, Smith ML, Rosenberg SH, Elmore SW, Tse C. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14(5):943–51. [PubMed: 17205078]
- 47.
- Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, Huang DC, Kile BT. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128(6):1173–86. [PubMed: 17382885]
- 48.
- Tunquist BJ, Woessner RD, Walker DH. Mcl-1 Stability Determines Mitotic Cell Fate of Human Multiple Myeloma Tumor Cells Treated with the Kinesin Spindle Protein Inhibitor ARRY-520. Mol. Cancer Ther. 2010;9(7):2046–2. [PubMed: 20571074]
- 49.
- Walensky LD, Stewart ML, Cohen N. Small Molecules for the Modulation of MCL-1 and methods of modulating cell death, cell division, cell differentiation, and methods of treating disorders. WO2011094708.
- 50.
- Stewart ML, Fire E, Keating AE, Walensky LD. The MCL-1 BH3 Helix is an Exclusive MCL-1 inhibitor and Apoptosis Sensitizer. Nat Chem Biol. 2010;6(8):595–601. [PMC free article: PMC3033224] [PubMed: 20562877]
- 51.
- Zhang Z, Wu G, Xie F, Song T, Chang X. 3-Thiomorpholin-8-oxo-8H-acenaphtho[1,2-b]pyrrole-9-carbonitrile (S1) Based Molecules as Potent, Dual Inhibitors of B-Cell Lymphoma 2 (Bcl-2) and Myeloid Cell Leukemia Sequence 1 (Mcl-1): Structure-Based Design and Structure-Activity Relationship Studies. J Med Chem. 2011;54(4):1101–1105. [PubMed: 21235240]
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- The imbalance between Bim and Mcl-1 expression controls the survival of human myeloma cells.[Eur J Immunol. 2004]The imbalance between Bim and Mcl-1 expression controls the survival of human myeloma cells.Gomez-Bougie P, Bataille R, Amiot M. Eur J Immunol. 2004 Nov; 34(11):3156-64.
- Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax).[Cell Death Dis. 2015]Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax).Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, Nimmer P, Jin S, Smith M, Xiao Y, et al. Cell Death Dis. 2015 Jan 15; 6(1):e1590. Epub 2015 Jan 15.
- A novel BH3 mimetic efficiently induces apoptosis in melanoma cells through direct binding to anti-apoptotic Bcl-2 family proteins, including phosphorylated Mcl-1.[Pigment Cell Melanoma Res. 2015]A novel BH3 mimetic efficiently induces apoptosis in melanoma cells through direct binding to anti-apoptotic Bcl-2 family proteins, including phosphorylated Mcl-1.Liu Y, Xie M, Song T, Sheng H, Yu X, Zhang Z. Pigment Cell Melanoma Res. 2015 Mar; 28(2):161-70. Epub 2014 Dec 18.
- Review Selective Bcl-2 Inhibitor Probes.[Probe Reports from the NIH Mol...]Review Selective Bcl-2 Inhibitor Probes.Zou J, Ardecky R, Pinkerton AB, Sergienko E, Su Y, Stonich D, Curpan RF, Simons PC, Zhai D, Diaz P, et al. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Mcl-1 inhibitors: a patent review.[Expert Opin Ther Pat. 2017]Review Mcl-1 inhibitors: a patent review.Chen L, Fletcher S. Expert Opin Ther Pat. 2017 Feb; 27(2):163-178. Epub 2016 Nov 17.
- ML311: A Small Molecule that Potently and Selectively Disrupts the Protein-Prote...ML311: A Small Molecule that Potently and Selectively Disrupts the Protein-Protein Interaction of Mcl-1 and Bim: A Probe for Studying Lymphoid Tumorigenesis - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...