

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

The cdc2-like (Clk) kinases are critical elements of several important regulatory pathways involving pre-mRNA splicing. These kinases act by phosphorylating the serine- and arginine-rich (SR) proteins, which direct the early events of spliceosome assembly necessary for proper mRNA maturation. Mutations causing splicing defects have been linked to a number of disease processes, making the search for small-molecule regulators of enzymes associated with spliceosome function important for both the further study and potential treatment of these disorders. The dual-specificity tyrosine-regulated (Dyrk) kinases are associated with numerous cellular processes and disorders. In particular, overexpression of Dyrk1A may contribute to Down syndrome neurodevelopmental abnormalities, and Dyrk1B/Mirk has been implicated in the cellular repair of cancer cells impaired by chemotherapy. Developing inhibitors of the Dyrk kinases would likely have important biochemical and therapeutic uses. Continuing in the search for selective inhibitors of Clk and Dyrk kinases, we now report a series of amino-substituted pyrimidines with excellent potencies against several Clk and Dyrk isoforms. The probe compound and its analogs have been subjected to Clk and Dyrk isoform selectivity profiling by Ambit Biosciences and three compounds have also been sent for KINOMEscan analysis to assess their selectivity in the context of the greater kinome. The reported compounds are low-nanomolar Clk and Dyrk inhibitors, with the most potent compound having IC50s <10 nM against Clk1, Clk4, Dyrk1A and Dyrk1B. The probe molecule (ML315) is a substituted aminopyrimidine and structurally distinct from previous quinazoline probes. ML315 should provide biochemical investigators with a new tool to be used in parallel with the quinazoline probes in efforts to determine which Clk and/or Dyrk isoforms are associated with particular biochemical events. Until isoform-selective inhibitors of each Clk and Dyrk kinase can be developed, parallel use of the available panel of inhibitors should enable biological investigations with these kinase families to proceed through a “process of elimination” approach.
Screening Center Name & PI: NIH Chemical Genomics Center, Christopher P. Austin
Chemistry Center Name & PI: University of Kansas Specialized Chemistry Center; Jeffrey Aubé
Assay Submitter & Institution: Tom Misteli, National Cancer Institute
PubChem Summary Bioassay Identifier (AID): 1997
Probe Structure & Characteristics

ML315
| CID/ML# | Target Name | IC50/EC50 (nM) [SID, AID | Anti-target Name(s) | IC50/EC50 (nM) [SID, AID] | Fold Selective | Secondary Assay(s) Name: IC50/EC50 (nM) [SID, AID] |
|---|---|---|---|---|---|---|
| CID-46926514/ML315 | Cdc2-like kinase 4 (Clk4) | 68 nM [SID 99431981, AID 624034] | Clk1 | 68 nM [SID 99431981, AID 624047] 231 nM [SID 99431981, AID 624048] >10,000 nM [SID 99431981, AID 624049] 282 nM [SID 99431981, AID 624045] 1156 nM [SID 99431981, AID 624046] | 1 fold | Kinase panel: Selectivity determined across the kinome via profiling within KinomeScan (DiscoveRx, http://www Additional activity against CSNK1E, MAP3K1, PKNB, PRKCE. |
| Clk2 | 3.4 fold | |||||
| Clk3 | >147 fold | |||||
| Dyrk1A | 4.1 fold | |||||
| Dyrk1B | 17 fold |
1. Recommendations for Scientific Use of the Probe
Post-transcriptional and post-translational processes are required to expand the 20,000–25,000 genes into the estimated 50,000–500,000 human protein variants.1 Alternate gene splicing is one mechanism by which this rich diversity of protein isoforms is achieved, and novel tools to help researchers understand and control gene splicing outcomes continue to be of considerable interest. Since the report of the first Clk inhibitor probe from this project (CID 44968231/ML167), there have been a limited number of reports on additional inhibitors of the Clk kinases. Each has a distinctive signature and ML315 represents a new chemotype with Clk activity and a unique structural and phamacological signature with which to interogate this complex process. Like the other inhibitors of this target, ML315 will be used to interrogate the role that the Clk kinases play in altering SR protein phosphorylation and the resulting consequences on gene splicing.
Apart from Clk4 (inhibited by ML167), no selective inhibitors have been reported for the other Clk isoforms (Clk1-Clk3); published Clk inhibitors either potently inhibit more than one isoform or have not been tested against all four isoforms. Using the available panel of inhibitors, including ML315, in parallel would allow biological investigations involving these kinases to proceed until isoform-selective inhibitors become available. One approach to using the three probes in parallel is described here: Using previous probe ML167 in Assay 1, one can inhibit Clk4 (136 nM) and observe the effect. Using previous probe ML106 in Assay 2, one can inhibit Clk1, Clk4, and Dyrk1A (all <100 nM) and observe the effect. Phenotypic or biochemical differences between Assay 1 and Assay 2 could be attributed to the lack of Clk1 or Dyrk1A activity. Using the new probe (ML315) in Assay 3, one can potently inhibit Clk1 and Clk4 (<100 nM), while only moderately inhibiting Clk2 and Dyrk1A (>200 nM). Phenotypic or biochemical differences between Assay 2 and Assay 3 could be attributed to the lack of Clk1 activity, allowing the roles Clk1 and Dyrk1A to be further distinguished. The differences observed between Assay 2 and Assay 3 might also be the result of moderate Clk2 inhibition. The parallel use of these probes is further discussed in Section 4.1. The previously reported probe (ML167) has been requested by numerous researchers and we plan to offer ML315 to researchers alongside this agent. We have additionally made other Clk inhibitors from the literature and offer each to researchers in the hopes that off-target activities associated with the divergent polypharmacology of each chemotype will be mitigated by parallel assessment of the activity of multiple Clk inhibitors.
2. Materials and Methods
2.1. Assays
Overall Assay Strategy
Both the quinazoline and pyrimidine scaffolds were originally identified from a cell-based assay aimed at identifying modulators of Lamin A splicing (AID 1487). Based upon these agents’ structures, we considered that kinase targets may be the mechanistic basis for the activity within this assay. From the literature it was determined that the cdc2-like kinases were likely targets and we followed up this phenotypic assay with an in vitro assay to determine these agents’ activity versus Clk4. In addition to this assay, we utilized commercially available assays from Reaction Biology (www.reactionbiology.com) and DiscoveRx (www.kinomescan.com/) to assess the selectivity of these agents.
Clk 4 qHTS Assays
The application of bioluminescence to ATPase assays has relied on a substrate depletion format. In these assays, the ATP dependence of firefly luciferase is used to measure the remaining ATP concentration, where the luminescence signal is inversely proportional to kinase activity.12–16 To provide a signal-to-background of approximately 2-fold, the substrate must be depleted by at least 50%. Operating enzyme assays under these high conversion conditions is not at all optimal for classical enzymological studies. However, this is acceptable for HTS, as shifts in potency are typically less than 2-fold with a percent conversion < 80%. Given that HTS assays typically show variability in potency determinations between ~2–3-fold, shifts due to high conversions in the range of 50–80% will not be easily discernible from the assay noise even if the assay is performed at lower conversions. Therefore, ATP depletion has become a popular choice to perform generic HTS assays for ATPases, particularly protein kinases.
We used two bioluminescent assays for Clk4 qHTS (Figure 1). Measurement of ATP depletion was assessed using the Kinase-Glo™ assay system (AID 1770), where a firefly luciferase detection reagent containing D-luciferin and buffer components are added to detect the remaining ATP following the Clk4 kinase assay (Figure 1a). The second system, ADP-Glo® measures kinase activity by quantifying the amount of ADP formed after the kinase reaction (AID 1771). Bioluminescent detection of ADP levels is achieved through the addition of two different detection reagents (Figure 1b). First, a reagent that stops the protein kinase reaction and depletes the remaining ATP is added. Then, a second reagent is added to stop ATP degradation. In addition, the second reagent also contains an enzyme such as pyruvate kinase that efficiently converts the ADP to ATP, and the same firefly luciferase/D-luciferin components present in Kinase-Glo, which generate a luminescent signal proportional to the ADP concentration produced. Therefore, the two assay formats show opposite luminescent signal changes in response to protein kinase inhibitors.

Figure 1
Bioluminescent assays used for Clk4 qHTS. a. Bioluminescent measurement of ATP depletion using Kinase-Glo. b. Bioluminescent measurement of ADP formation using ADP-Glo.
All compounds were screened using a qHTS approach,15 in which compounds were assayed using at least seven concentrations to generate concentration-response curves for each compound tested. The methodology for creating a concentration-titration series between successive copies of library plates for the purpose of large-scale titration-based screening has been described. Briefly, qHTS uses an inter-plate dilution method where the first plate contains the highest concentration of a set of compounds in DMSO, while subsequent plates contain the same compounds in the same well locations, but at successively lower concentrations. Using the protocol outlined above, we calculated a plate throughput of 18 plates/hr, or approximately 7 samples/sec, on the Kalypsys robotic system, which means that a 7 point CRC was obtained every second on the robotic system.
Table 1Assay protocol: The optimized 1536-well protocol
| Kinase-Glo | ADP-Glo | |||||
|---|---|---|---|---|---|---|
| Step | Parameter | Value | Description | Parameter | Value | Description |
| 1 | Reagent | 2 μL | ATP/peptide | Reagent | 2 μL | ATP/peptide |
| 2 | Library | 23 nL | 0.5 nM- 46 μM | Library | 23 nL | 0.6 nM- 55.2 μM |
| 3 | Controls | 23 nL | TG003 | Controls | 23 nL | TG003 |
| 4 | Reagent | 1 μL | Clk4 | Reagent | 0.5 μL | Clk4 |
| 5 | Time | 4.5 hrs | r.t. incubation | Time | 1 hr | r.t. incubation |
| 6 | Reagent | 3 μL | Kinase-Glo | Reagent | 2.5 μL | Deplete ATP |
| 7 | Read | 2 sec | ViewLux | Time | 45 min | r.t. incubation |
| 8 | Reagent | 5 μL | ADP→ATP/Luc | |||
| 9 | Time | 30 min | r.t. incubation | |||
| 10 | Read | 2 sec | ViewLux | |||
| Step | Notes | Notes | ||||
| 1 | 100 μM RS peptide, 1 μM ATP (final) concentration in buffer; FRD dispense | 100 μM RS peptide, 1 μM ATP (final) concentration in buffer; FRD dispense | ||||
| 2 | Pin-tool transfer compound library for a (final) range of 46 μM to 0.5 nM | Pin-tool transfer compound library for a (final) range of 55.2 μM to 0.6 nM | ||||
| 3 | ||||||
| 4 | Clk4 at 25 nM final, FRD dispense | Clk4 at 25 nM final, FRD dispense | ||||
The assay showed excellent performance (the signal-to-background ratio was 3.2 +/− 0.07, the average Z′ screening factor associated with each plate was 0.86 +/− 0.02 and the CV was 7.2 +/− 1.9, indicating a robust performance of the screen).
Clk4 confirmation assay
Kinase-Glo™ assay to measure ATP depletion was used in the confirmation of Clk4 inhibition. All compounds were tested in 12-pt concentration response from 77 μM to 0.4 nM.
Dyrk1A selectivity assay
Among the members of the CMGC group, the Clks and the Dyrks are very closely related. Therefore to determine the activity and selectivity of the Clk4 pyrimidine analogs, a bioluminescent counter-screen against Dyrk1A was developed. Dyrk1A (Invitrogen, cat # PV3785) was assayed using ATP and DYRKtide (AnaSpec, cat# 62698) as substrates. Promega Kinase-Glo (cat# V6712) technology was used to detect the residual ATP following kinetic reaction. The Kinase-Glo Plus contains Ultra-Glo luciferase and D-luciferin which generates a bioluminescence signal from the remaining ATP. No enzyme and harmine (Sigma, cat # 286044-1G), a compound shown to inhibit Dyrks, were used as positive controls; DMSO treatment was used as a negative control. All compounds were tested in 12-pt concentration response from 77μM to 0.4nM.
Dyrk1A assay protocol summary: Two μL/well of substrate solution (8 μM Dyrktide, 2 μM ATP, 25 mM Tris-HCl pH 7.5, 10 mM MgCl2, 0.5 mM EGTA, 5 mM cystein, 0.01% Triton X-100 final concentration) was dispensed into 1536-well assay plates (Greiner, solid white, medium-binding plates) with Aurora Discovery BioRAPTR Flying Reagent Dispenser (FRD, Beckton-Dickenson). Compound solution (23 nL) was transferred to the assay plate using a Kalypsys pin tool equipped with a 1536-pin tool. One μL/well of enzyme solution (5 nM Dyrk1A, 2 5mM Tris-HCl pH7.5, 10 mM MgCl2, 0.5 mM EGTA, 5 mM cystein, 0.01% Triton X-100 final concentration) was then added using FRD, yielding a total kinase reaction volume of 3 μL/well. After 2 hours of room temperature incubation, 3 μL of Kinase-Glo detection reagent was added for a final assay volume of 6 μL/well. The plates were spun for 30 seconds (1000 rpm) and incubated at ambient temperature for 2 minutes, followed by a luminescence read using Perkin Elmer ViewLux plate reader at 3 seconds exposure time and 2× binning.
Clk1-4, Dyrk1A, Dyrk1B Assay
Reaction Biology (www.reactionbiology.com) offers in vitro assays for multiple kinases including all Clk isoforms, Dyrk1A and Dyrk1B. The assay format relies upon a 33P-γ-ATP radiometric based filtration binding assay. Please refer to BioAssay AID 624047, AID 624048, AID 624049, AID 624034, AID 624045 and AID 624046.
KinomeScan Assay
The KinomeScan assay [originally developed by Ambit Biosciences and currently offered by DiscoverX (www.kinomescan.com/)] is based upon a competition binding assay of kinases fused to a proprietary tag.
2.2. Probe Chemical Characterization
Probe Chemical Structure, Physical parameters, and Properties
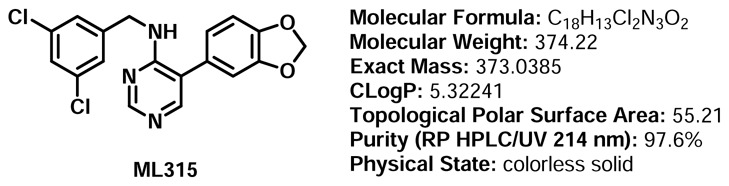
Structure Verification and Purity: 1H NMR, 13C NMR, RP HPLC/UV/HRMS Data
Proton and carbon NMR data for ML315: Detailed analytical methods and associated instrumentation are described in section 2.3, entitled “Probe Preparation”, under general experimental and analytical details. The numerical experimental proton and carbon NMR data are presented.
Proton NMR Data for ML315:1H NMR (500 MHz, DMSO-d6): δ 8.41 (s, 1H), 7.96 (s, 1H), 7.41 (t, J = 2.0 Hz, 1H), 7.35-7.32 (m, 3H), 7.05 (d, J = 8.0 Hz, 1H), 7.02 (d, J = 1.5 Hz, 1H), 6.90 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 6.09 (s, 2H), 4.54 (d, J = 6.5 Hz, 2H).
Carbon NMR Data for ML315:13C NMR (125 MHz, DMSO-d6): δ 158.9, 156.9, 153.4, 147.8, 147.3, 144.7, 133.8 (2), 127.6, 126.2, 125.9 (2), 122.6, 119.1, 109.4, 109.0, 101.3, 42.8.
RP HPLC/UV/HRMS Data for ML315: Detailed analytical methods and associated instrumentation are described in section 2.3, entitled “Probe Preparation”, under general experimental and analytical details. Purity assessment by RP HPLC/UV/HRMS at 214 nm for ML315 revealed purity of 97.6% (retention time = 3.37 minutes). HRMS (m/z): calcd for C18H14Cl2N3O2 (MH+) 374.0465; found 374.0486.
Solubility
Aqueous solubility for the probe was assessed in phosphate-buffered saline (PBS) at room temperature. PBS by definition is 137 mM NaCl, 2.7 mM KCl, 10 mM sodium phosphate dibasic, 2 mM potassium phosphate monobasic at a pH of 7.4 (ref to SBMRI). Under these conditions, the solubility of ML315 was determined to be 1.15 μM. Solubility was also assessed in the assay media of the primary assay (25 mM Tris pH7.5, 10 mM MgCl2, 0.5 mM EGTA, 2.5 mM DTT, 0.01% Triton X-100). Under these conditions, the solubility of probe ML315 was determined to be 1.71 μM. Though the solubility of ML315 is limited, the compound concentration was 17-fold greater in PBS than the measured IC50 as determined in the AMBIT panel, and in the assay media, the compound concentration was 25-fold greater than the measured IC50, allaying concerns about solubility-limited potency and SAR.
Stability
Aqueous stability for the probe was assessed using two solvent systems (100% aqueous PBS, and 50:50 aqueous PBS:acetonitrile). The probe’s stability was measured in aqueous PBS (no antioxidants or other protectants, DMSO concentration below 1%, room temperature) and the results are reported as circles in the graph in Figure 2. The probe’s stability was also measured in 50:50 aqueous PBS and acetonitrile and the results are reported as triangles in the graph in Figure 2. Stability data in each case is depicted as the loss of compound with time over 48 hours with a minimum of six time points and providing the percent compound remaining after 48 hours. The apparent compound instability in aqueous PBS is much more likely attributed to compound insolubility in that medium, because 100% of the compound was detected after 48 hours in PBS with 50% acetonitrile (50% ACN).

Figure 2
Stability studies for ML315 in aqueous PBS (circles) and 50:50 aqueous PBS:acetonitrile (triangles).
Synthesis Route
The probe compound ML315 was synthesized according to Scheme 1. Bromination of 4-aminopyrimidine afforded 5-bromo-4-amino pyrimidine, which was subjected to Suzuki cross-coupling conditions with 3,4-(methylenedioxy)phenylboronic acid in a microwave reactor to produce S-1. Reductive amination of S-1 with 3,5-dichlorobenzaldehyde delivered ML315, which was purified by preparative reverse-phase HPLC.

Scheme 1
Synthetic route to ML315.
Benzodioxole and benzodioxane analogs were synthesized via the sequence shown in Scheme 2. Suzuki cross-coupling of 2-amino-4-chloropyrimidine with either 3,4-(methylenedioxy)phenylboronic acid or 1,4-benzodioxane-6-boronic acid in a microwave reactor afforded S-2 or S-3, respectively. Subjecting the cross-coupling products to reductive amination conditions with the appropriate aldehyde in each case produced the desired analogs, which were purified by preparative reverse-phase HPLC.

Scheme 2
Synthetic route to oxygenated analogues.
Indazole analogs were synthesized according to the sequence shown in Scheme 3. 2,4-Dichloropyrimidine was reacted with 3,4-dichlorobenzylamine via SNAr reaction in a microwave reactor at 110 °C, producing S-4 and S-5 in a ratio of 72:28. The isomeric mixture was readily separated using silica gel column chromatography. The individual isomers S-4 and S-5 were then used in microwave-assisted Suzuki cross-coupling reactions with the appropriate indazolylboronic acids to afford the desired indazole analogs, which were purified by preparative reverse-phase HPLC. These compounds were also synthesized via Suzuki cross-coupling/reductive amination sequences starting from either 2-amino-4-chloropyrimidine or 4-amino-2-chloropyrimidine to confirm the identities of the desired analogs.
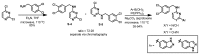
Scheme 3
Synthetic route to indazole analogues.
Submission of Probe and Five Supporting Analogues to the MLSMR
Samples of the probe and five analogs were prepared, analytically characterized, and shipped to the MLSMR. The structures for the five supporting analogues are shown in Table 2. The screening results from NCGC in-house bioluminescent luciferase-based assays are shown in parentheses, while the remaining data (IC50 values, nM) was generated using a commercial vendor for AMBIT subtype selectivity profiling against the Clk and Dyrk kinases.
Table 2
Supporting analogs with IC50 values (nM) against Clk and Dyrk kinases.
2.3. Probe Preparation
General experimental and analytical details
All reagents were used as received from commercial suppliers. Microwave reactions were carried out using a Biotage Initiator. The 1H and 13C spectra were recorded on Bruker Avance 400 MHz or 500 MHz spectrometers. Chemical shifts are reported in parts per million and referenced to the center line of residual solvent signals: CDCl3 (7.26 ppm for 1H and 77.23 ppm for 13C), d3-MeCN (1.94 ppm for 1H and 1.39 ppm for 13C), and d6-DMSO (2.50 ppm for 1H and 39.51 ppm for 13C). Flash column chromatography separations were performed using Sorbent Technologies standard grade silica gel (40–63 μm particle size, 230 × 400 mesh) with compressed nitrogen as a source of positive pressure or the Teledyne Isco CombiFlash RF using RediSep RF silica gel columns. TLC was performed on Analtech UNIPLATE silica gel GHLF plates (gypsum inorganic hard layer with fluorescence). TLC plates were developed using UV light or aqueous KMnO4. HPLC/MS analysis was carried out with gradient elution (5% CH3CN to 100% CH3CN) on an Agilent 1200 RRLC with a photodiode array UV detector and an Agilent 6224 TOF mass spectrometer (also used to produce high resolution mass spectra). Purification was carried out by mass-directed fractionation with gradient elution (a narrow CH3CN gradient was chosen based on the retention time of the target from LCMS analysis of the crude sample) on an Agilent 1200 instrument with photodiode array detector, an Agilent 6120 quadrupole mass spectrometer, and a HTPAL LEAP autosampler. Fractions were triggered using an MS and UV threshold determined by HPLC/MS analysis of the crude sample. The conditions for HPLC analysis included the following: Waters BEH C-18, 1.7 μm, 2.1 × 50mm column; 0.6 ml/min flow rate; and pH 9.8 NH4OH aqueous mobile phase. The conditions for purification included: Waters XBridge C18 5μm, 19 × 150mm column; 20 ml/min flowrate pH 9.8 NH4OH aqueous mobile phase.
The probe was prepared using the following protocols
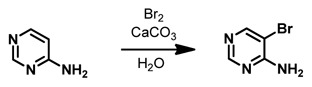
5-Bromopyrimidin-4-amine. This compound was synthesized according to the published procedure.17 4-Aminopyrimidine (1.00 g, 10.5 mmol, 1.0 equiv) and CaCO3 (263 mg, 2.63 mmol, 0.25 equiv) were combined in H2O (20 mL) and Br2 (1.06 mL, 20.5 mmol, 2.0 equiv) was added dropwise. The reaction mixture was stirred at 60 °C for 45 minutes, cooled to room temperature, and then poured into CH2Cl2 (20 mL). The layers were separated and the aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were set aside. The aqueous layer was treated with 20% aqueous K2CO3 until pH 9–10 was achieved, and the product precipitated. The precipitate was removed via filtration and dried, affording the product (1.08 g, 6.22 mmol, 59%) as a light tan solid. 1H NMR (500 MHz, DMSO-d6): δ 8.32 (s, 2H), 7.87–6.62 (v br s, 2H). 13C NMR (125 MHz, DMSO-d6): δ 160.1, 156.8, 156.0, 102.5.
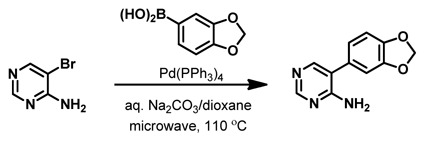
5-(Benzo[d][1,3]dioxol-5-yl)pyrimidin-4-amine. 5-Bromopyrimidin-4-amine (400 mg, 2.30 mmol, 1.0 equiv) was dissolved in dioxane (7 mL) in a 20-mL microwave vial and to it was added 3,4-(methylenedioxy)-phenylboronic acid (763 mg, 4.60 mmol, 2.0 equiv), aqueous Na2CO3 (2.0 M, 3.45 mL, 6.90 mmol, 3.0 equiv), and Pd(PPh3)4 (266 mg, 0.23 mmol, 0.10 equiv). The sealed vial was placed in a microwave reactor and heated at 110 °C for 30 minutes. The crude reaction mixture was partitioned between CH2Cl2 (20 mL)/H2O (20 mL) and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 20 mL), and the combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The crude reaction mixture was purified via silica gel column chromatography using MeOH/CH2Cl2 (1:19), affording the product (220 mg, 1.02 mmol, 44%) as a light tan solid. 1H NMR (500 MHz, DMSO-d6): δ 8.33 (s, 1H), 7.95 (s, 1H), 7.00 (d, J = 8.0 Hz, 1H), 6.97 (d, J = 1.5 Hz, 1H), 6.87 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 6.60 (br s, 2H), 6.06 (s, 2H) 13C NMR (125 MHz, DMSO-d6): δ 160.8, 157.0, 154.0, 147.7, 147.0, 128.3, 122.2, 117.7, 109.0, 108.9, 101.2.

5-(Benzo[d][1,3]dioxol-5-yl)-N-(3,5-dichlorobenzyl)pyrimidin-4-amine. 5-(Benzo[d][1,3]dioxol-5-yl)pyrimidin-4-amine (150 mg, 0.70 mmol, 1.0 equiv) was suspended in MeCN (15 mL), and then 3,5-dichlorobenzaldehyde (610 mg, 3.49 mmol, 5.0 equiv), ClTi(Oi-Pr)3 (95%, 1.05 mL, 4.20 mmol, 6.0 equiv) and AcOH (5 drops) were added. The reaction mixture was stirred for 5 minutes at room temperature and then Na(OAc)3BH (95%, 779 mg, 3.49 mmol, 5.0 equiv) was added. The mixture was stirred for two hours and then aqueous NH4OH (15%, 15 mL) was added. The mixture was stirred for a further one hour and then solids were filtered off and washed with CH2Cl2 (20 mL) and H2O (20 mL). The layers of the biphasic mixture were separated and the aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were dried over Na2SO4 and then concentrated under reduced pressure. The crude reaction mixture was submitted for preparative reverse-phase HPLC purification, affording the product as a white solid (86.1 mg, 0.23 mmol, 33%). 1H NMR (500 MHz, DMSO-d6): δ 8.41 (s, 1H), 7.96 (s, 1H), 7.41 (t, J = 2.0 Hz, 1H), 7.35-7.32 (m, 3H), 7.05 (d, J = 8.0 Hz, 1H), 7.02 (d, J = 1.5 Hz, 1H), 6.90 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 6.09 (s, 2H), 4.54 (d, J = 6.5 Hz, 2H). 13C NMR (125 MHz, DMSO-d6): δ 158.9, 156.9, 153.4, 147.8, 147.3, 144.7, 133.8 (2), 127.6, 126.2, 125.9 (2), 122.6, 119.1, 109.4, 109.0, 101.3, 42.8. HRMS (ESI/APCI) Calcd for C18H14Cl2N3O2 ([M+H]+): 374.0465. Found: 374.0486.
3. Results
3.1. Dose Response Curves for Probe

Figure 3Dose response curve for ML315
3.2. Cellular Activity
We are currently conducting studies with ML315 (alongside ML106 and ML167) to assess all of these agents’ ability to modulate the splicing of VEGF isoforms (namely VEGFa and VEGFb) via a cell-based RT-PCR assay. Preliminary results suggest that these agents are capable of modulating VEGF splice variants.
3.3. Profiling Assays
In vitro pharmacokinetics profiling
The in vitro pharmacokinetic (PK) properties of the probe (ML315) were profiled using a standard panel of assays (Table 3). The results from this profile suggested that ML315 has modest aqueous and assay media solubility, good permeability across an artificial membrane (PAMPA), good plasma stability but low microsomal stability.
Table 3
Summary of in vitro ADME properties of ML315.
Broad-spectrum target profiling: ML315 was submitted for assessing off-target pharmacology using a Ricerca LeadProfiling® screen made up of 67 assays. The probe was assayed in duplicate at a concentration of 10 μM for all targets, and the following responses were noted as ≥ 50% inhibition or stimulation for biochemical assays (see Table 4).
Table 4
Off-target pharmacology data for ML315 highlighting targets with ≥ 50% inhibition or stimulation.
4. Discussion
4.1. Comparison to Existing Art and How the New Probe is an Improvement
The new probe (ML315, SID 99431981, CID 46926514, 91) has a pyrimidine core, which is a chemotype that is structurally distinct from prior probes (ML106 and ML167) having quinazoline cores. ML315 exhibits a 1,2-relationship between the substituents appended to the pyrimidine ring, while ML106 and ML167 display appended substituents on different rings of the fused bicyclic quinazoline. Though benzodioxolane substituents are found in both ML315 and ML106, the benzodioxolane is directly attached to the pyrimidine in ML315, while it is bound to the benzene ring in ML106, providing these inhibitors with different molecular topologies. For instance, comparing the distances between the NH groups and oxygen atoms of the benzodioxolanes in each inhibitor, it is clear that these hydrogen-bonding groups are spread much further apart in ML106 than in ML315. The divergent topological features of ML315 and ML106, including the differences in arrangement of hydrogen-bond-capable functional groups, is expected to distinguish these probes from one another during binding events in both the Clk/Dyrk families and throughout the greater kinome, as each will fit differently into the binding pocket of a given enzyme. Furthermore, the effect of the benzodioxolane’s electron density on the basicity of the benzylamine is also expected to differ between the pyrimidine and quinazoline chemotypes. In the quinazoline (ML106), electron density is spread over the six additional atoms of the benzene ring, diminishing the effect on the benzylamine appended to the fused pyrimidine. However, in the pyrimidine (ML315), the electron-rich benzodioxolane and the benzylamine are both bound to the same heterocycle.
Although ML167 (Entry 1 in Table 5) selectively inhibits Clk4 (136 nM), none of the remaining Clk inhibitors appear to have single isoform selectivity (> 10-fold activity) against Clk1-Clk3; published Clk inhibitors either potently inhibit more than one isoform or have not been tested against all four isoforms. Therefore, until single-isoform-selective inhibitors can be developed, the parallel use of available inhibitors can still be used to gather biological data involving these kinases. The selectivity profile for the new probe differs from the profiles of previous probes in interesting and potentially useful ways. As mentioned above, ML167 is an isoform-selective Clk4 inhibitor. Comparing entries 2 and 3, it may be seen that while this prior quinazoline probe ML106 is a potent inhibitor of Clk1, Clk4, and Dyrk1A and not very potent against Clk2, the new pyrimidine probe ML315 is a potent inhibitor of Clk1 and Clk4 and a moderate inhibitor of Clk2 and Dyrk 1A. Using ML315 in parallel with previous probes ML106 and ML167 should allow researchers to address questions that would not be possible with the previous probes alone. For instance, employing ML167 in a biochemical assay (Assay 1) would allow one to determine the effect of inhibiting Clk4 (136 nM). Administering ML106 (Assay 2) would then allow one to inhibit Clk1, Clk4, and Dyrk1A (59 nM, 39 nM, and 62 nM, respectively). Differences between the results of Assay 1 and Assay 2 might be attributed to the lack of Clk1 and Dyrk1A activity, thereby helping to define the functions of these two kinases. Running the assay a third time (Assay 3) using ML315 would further allow one to inhibit Clk1 (68 nM) and Clk4 (68 nM), while only moderately inhibiting Dyrk1A (282 nM) and Clk2 (231 nM). Differences between the results of Assay 2 and Assay 3 might in this case be attributed to the loss of Clk1 activity, allowing the functions of Clk1 and Dyrk1A to be further separated and understood. These differences might also result from the moderate inhibition of Clk2. Thus, by running the same assay with each of the three probes, researchers should be able to exclude isoforms from consideration and begin to hone in on which Clk or Dyrk kinase is responsible for particular biochemical observations. Furthermore, in advanced pharmacological studies, compounds with similar activity profiles could have distinctly different liabilities (metabolic instability, toxicity, etc.), making the availability of multiple chemotypes with different chemical structures advantageous.
Table 5
IC50 values (nM) against Clk and Dyrk kinases for quinazoline and pyrimidine chemotypes.
5. References
- 1.
- Uhlen M, Ponten F. Antibody-based Proteomics for Human Tissue Profiling. Molecular & Cellular Proteomics. 2005;4(4):384–393. [PubMed: 15695805]
- 2.
- Prasad J, Manley JL. Mol Cell Bio. 2003;23:4139–4149. [PMC free article: PMC156123] [PubMed: 12773558]
- 3.
- Mott BT, Tanega C, Shen M, Maloney DJ, Shinn P, Leister W, Marugan JJ, Inglese J, Austin CP, Mistelli T, Auld DS, Thomas CJ. Bioorg Med Chem Lett. 2009;19:6700–6705. [PMC free article: PMC2807730] [PubMed: 19837585]
- 4.
- Rosenthal AS, Tanega C, Shen M, Mott BT, Bougie JM, Nguyen D.-T, Misteli T, Auld DS, Maloney DJ, Thomas CJ. Bioorg Med Chem Lett. 2011;21:3152–3158. [PMC free article: PMC3085634] [PubMed: 21450467]
- 5.
- Muraki M, Ohkawara B, Hosoya T, Onogi H, Koizumi J, Koizumi T, Sumi K, Yomoda J-i, Murray MV, Kimura H, Furuichi K, Shibuya H, Drainer AR, Suzuki M, Hagiwara M. J Biol Chem. 2004;279:24246–24254. [PubMed: 15010457]
- 6.
- Hekimi S, McBride K, Hihi AK, Kianicka I, Wang Y, Hayes SL, Guimond M.-P, Sevigny G, Dumas D, Smith J. WO 2008014602.
- 7.
- Fedorov O, Huber K, Eisenreich A, Filippakopoulos P, King O, Bullock AN, Szklarczyk D, Jensen LJ, Fabbro D, Trappe J, Rauch U, Bracher F, Knapp S. Chem Biol. 2011;18:67–76. [PMC free article: PMC3145970] [PubMed: 21276940]
- 8.
- Debdad M, Carreaux F, Renault S, Soundararajan M, Fedorov O, Filippakopoulos P, Lozach O, Babault L, Tahtouh T, Baratte B, Ogawa Y, Hagiwara M, Eisenreich A, Rauch U, Knapp S, Meijer L, Bazureau J.-P. J Med Chem. 2011;54:4172–4186. [PubMed: 21615147]
- 9.
- Gockler N, Jofre G, Papadopoulos C, Soppa U, Tejedor FJ, Becker W. FEBS Journal. 2009;276:6324–6337. [PubMed: 19796173]
- 10.
- Wang S, Wood G, Duncan KW, Meades C, Gibson D, McLachlan JC, Perry A, Blake D, Zheleva DI, Fischer P. PCT Int. Appl. 2005. p. 105. CODEN: PIXXD2 WO 2005042525 A1 20050512 CA 142:463748 AN 2005:409513 CAPLUS.
- 11.
- Aubé J, Thomas C, Schoenen FJ, Auld D, Coombs TC, Shen M, Tanega CUS. Provisional Application entitled “CLK and DYRK Inhibitors and Methods of Use Thereof” Serial No : 61/584,935. Filing Date: January 10, 2012.
- 12.
- Fan F, Wood KV. Assay Drug Dev Technol. 2007;5:127–36. [PubMed: 17355205]
- 13.
- Schroter T, Minond D, Weiser A, Dao C, Habel J, Spicer T, Chase P, Baillargeon P, Scampavia L, Schurer S, Chung C, Mader C, Southern M, Tsinoremas N, LoGrasso P, Hodder P. J Biomol Screen. 2008;13:17–28. [PubMed: 18227223]
- 14.
- Singh P, Harden BJ, Lillywhite BJ, Broad PM. Assay Drug Dev Technol. 2004;2:161–9. [PubMed: 15165512]
- 15.
- Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Proc Natl Acad Sci USA. 2006;103:11473–8. [PMC free article: PMC1518803] [PubMed: 16864780]
- 16.
- Tanega C, Shen M, Mott BT, Thomas CJ, MacArthur R, Inglese J, Auld DS. Assay & Drug Dev Tech. 2010;7:606–614. [PMC free article: PMC3096547] [PubMed: 20059377]
- 17.
- Reigan P, Gbaj A, Stratford IJ, Bryce RA, Freeman S. Eur J Med Chem. 2008;43:1248–1260. [PubMed: 17870212]
- 18.
- Woolard J, Wang W.-Y, Bevan HS, Qiu Y, Morbidelli L, Pritchard-Jones RO, Cui T.-G, Sugiono M, Waine E, Perrin R, Foster R, Digby-Bell J, Shields JD, Whittles CE, Mushens RE, Gillatt DA, Ziche M, Harper SJ, Bates DO. Cancer Res. 2004;64:7822–7835. [PubMed: 15520188]
- PMCPubMed Central citations
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- Small-molecule pyrimidine inhibitors of the cdc2-like (Clk) and dual specificity tyrosine phosphorylation-regulated (Dyrk) kinases: development of chemical probe ML315.[Bioorg Med Chem Lett. 2013]Small-molecule pyrimidine inhibitors of the cdc2-like (Clk) and dual specificity tyrosine phosphorylation-regulated (Dyrk) kinases: development of chemical probe ML315.Coombs TC, Tanega C, Shen M, Wang JL, Auld DS, Gerritz SW, Schoenen FJ, Thomas CJ, Aubé J. Bioorg Med Chem Lett. 2013 Jun 15; 23(12):3654-61. Epub 2013 Mar 30.
- Review An inhibitor of the Cdc2-like kinase 4 (Clk4).[Probe Reports from the NIH Mol...]Review An inhibitor of the Cdc2-like kinase 4 (Clk4).Rosenthal AS, Tanega C, Shen M, Mott BT, Bougie JM, Nguyen DT, Misteli T, Auld DS, Maloney DJ, Thomas CJ. Probe Reports from the NIH Molecular Libraries Program. 2010
- Potent and selective small molecule inhibitors of specific isoforms of Cdc2-like kinases (Clk) and dual specificity tyrosine-phosphorylation-regulated kinases (Dyrk).[Bioorg Med Chem Lett. 2011]Potent and selective small molecule inhibitors of specific isoforms of Cdc2-like kinases (Clk) and dual specificity tyrosine-phosphorylation-regulated kinases (Dyrk).Rosenthal AS, Tanega C, Shen M, Mott BT, Bougie JM, Nguyen DT, Misteli T, Auld DS, Maloney DJ, Thomas CJ. Bioorg Med Chem Lett. 2011 May 15; 21(10):3152-8. Epub 2011 Mar 4.
- The Nitro Group Reshapes the Effects of Pyrido[3,4-g]quinazoline Derivatives on DYRK/CLK Activity and RNA Splicing in Glioblastoma Cells.[Cancers (Basel). 2024]The Nitro Group Reshapes the Effects of Pyrido[3,4-g]quinazoline Derivatives on DYRK/CLK Activity and RNA Splicing in Glioblastoma Cells.Borisevich SS, Aksinina TE, Ilyina MG, Shender VO, Anufrieva KS, Arapidi GP, Antipova NV, Anizon F, Esvan YJ, Giraud F, et al. Cancers (Basel). 2024 Feb 19; 16(4). Epub 2024 Feb 19.
- Review A high-throughput screen for pre-mRNA splicing modulators.[Probe Reports from the NIH Mol...]Review A high-throughput screen for pre-mRNA splicing modulators.Auld D, Shen M, Thomas C. Probe Reports from the NIH Molecular Libraries Program. 2010
- Identification of selective inhibitors of cdc2-like kinases 1 and 4 (Clk1, Clk4)...Identification of selective inhibitors of cdc2-like kinases 1 and 4 (Clk1, Clk4) - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...