

Submitting Mitochondrial and Chloroplast Genomes to GenBank
Introduction
This document guides first timers through the entire process of submitting organelle genome(s) to GenBank. It also provides help for the submitters who were asked to resubmit their data because of lacking or inaccurate annotation. If you are already familiar with the submission process but you need to fix your annotation, go to the following sections of the document:
Organelle Genome Submission Path
The Features Step/Annotation with the Five-Column Feature Table
Troubleshooting Annotation/Feature Table Format
Organelle Genome Submission Path
Use BankIt to submit complete or incomplete organelle genome(s) to GenBank:
-
On the BankIt web page select the following radio button:
-
Sequence data not listed above (through BankIt): mRNA, genomic DNA, organelle, ncRNA, plasmids, other viruses, phages, synthetic constructs
-
Click the Start button (sign in to NCBI if needed).
-
On the resulting page, select the Start BankIt Submission button.
You can submit more than one organelle genome with one BankIt. However, you may need to separate your data into several BankIt submissions if you have numerous and/or large genomes. We cannot provide a hard limit for the data size of one BankIt as it will depend on your connection speed. If you get a server error while working in BankIt, your data size may be too large. You can use the tbl2asn command line software as the alternative tool for large data. You would use the same data input files (same formats) as in BankIt.
GenBank does not require reads from next generation sequencing technologies to accompany the assembled organelle genome submissions. However, you can submit your reads to Sequence Reads Archive (SRA) if that is necessary for your project. Please refer to the SRA submission guide. Once your SRA and GenBank records are processed, GenBank indexers can link your GenBank accessions with your SRA data.
Submit organelle genomes through the Genome Submission Portal ONLY if they accompany organism's nuclear genome submission.
Working with BankIt
BankIt is a web-based application comprised of several steps that include: Contact, Reference, Sequencing Technology, Nucleotide, Submission Category, Source Modifiers, Features, and Review and Correct. You will find comprehensive instructions within BankIt at each step. Here, we selectively provide instructions for three of the steps: Nucleotide, Source Modifiers, and Features. We are emphasizing parts that are relevant to organelle genome submissions.
Uploading genome sequence at the Nucleotide step
The Nucleotide page contains a section where you mark your genome(s) as complete, circular genomic DNA:

On the same page, you will upload your sequence(s) in the FASTA format (not aligned) or in an alignment format such as FASTA+GAP or NEXUS. All the formats are described/explained at the step in BankIt. Here, we illustrate a plain text file that contains three genomes (the sequences are truncated for illustration purpose):

Each FASTA starts with the ">"symbol followed by a sequence identifier (Sequence_ID). You can choose your own Sequence_IDs but keep them simple. Inclusion of the organism name within the square brackets allows for proper processing of the name from the FASTA definition line. You can follow the organism name by a descriptive text. It is important that you present the entire FASTA definition in a single line and then follow by a hard return before the sequence.
GenBank indexers will edit the definition lines in the published records according to GenBank standards, so you should not be overly concerned about matching your definition line to those in the database.
Describing sequence source at the Source Modifiers Step
The Source Modifiers page contains the Organelle/Location drop-down menu where you mark the organelle genome that you have:

Providing source modifiers using the web form
Choose applicable source modifiers that allow you to describe your sample from the source modifier pull down menu. Provide values as you have them for your sample. Commonly applicable source modifiers for organelle genomes are strain/isolate and/or voucher, geo_loc_name, and isolation_source. Here, we illustrate an example with detailed source information for the mitochondrial genome of Chinese short-limbed skink. The left hand-side of the image below (blue background) shows the selected modifiers and values as the submitter would have provided in BankIt. The right hand-side (yellow background) reflects the information in the source section of the processed GenBank record:
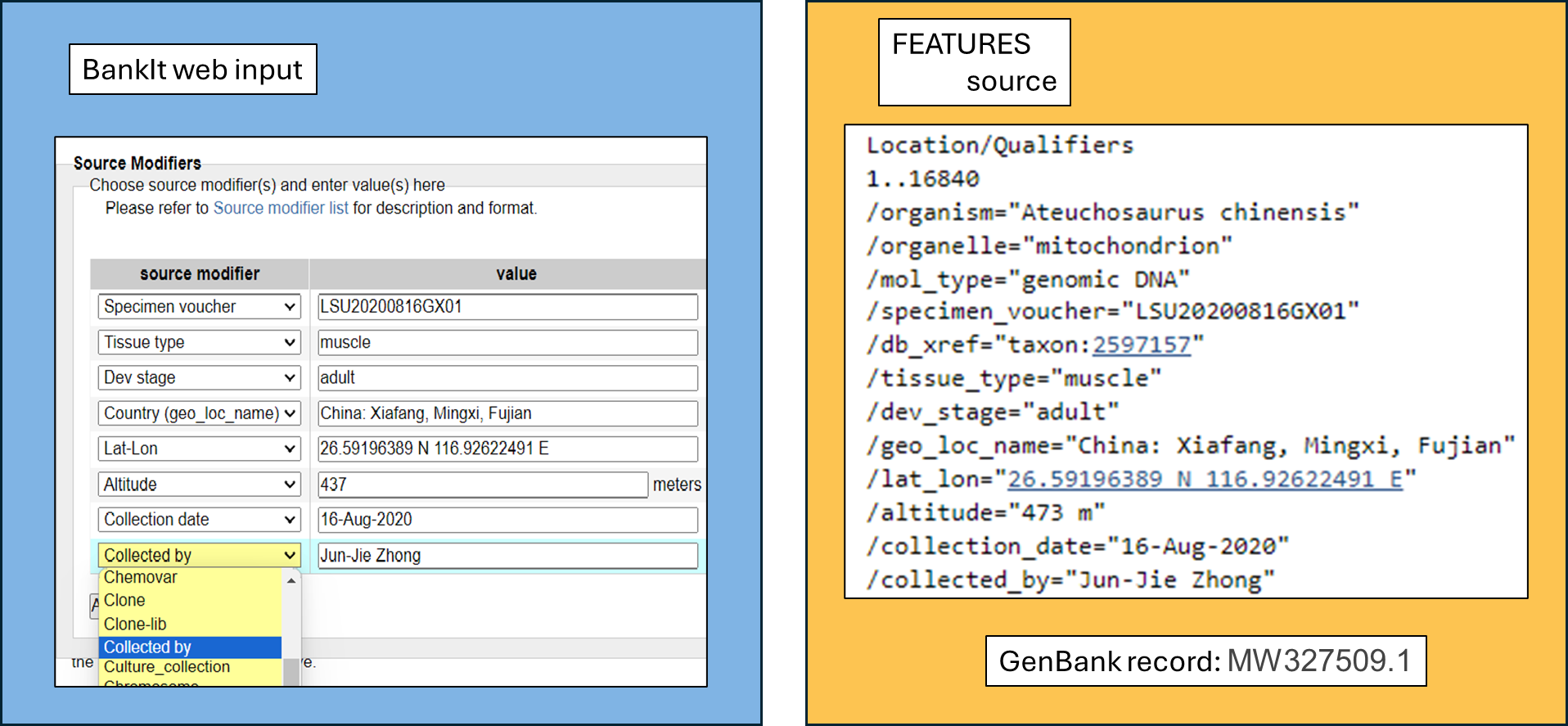
Note that GenBank indexers (not the submitter) provide some parts of source information (such as the Taxonomy db_xref ).
Providing source modifiers using a table (file input)
If you are submitting more than one genome and you need to provide different source values for each genome, you need to provide a source table (file input). The web form will not work in such case.
Provide unique identifiers to differentiate between the samples of the same species. For example, use isolate and provide the alphanumerical codes that you used in your study to differentiate between your samples. Other source modifiers that can serve as unique identifiers include strain, clone, culture_collection, and specimen_voucher.
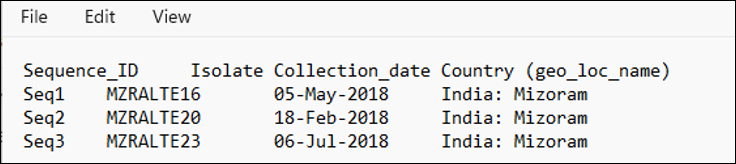
The source table for our example of the three Physalis chloroplast genomes shows unique isolate designations, different collection dates, and the same geographic location name/region. Note the first column of the table contains Sequence_IDs that match those in the FASTA file.
To format a source table, you can work with a spreadsheet:

Once completed, save the table as plain-text, tab delimited file (you cannot upload a spreadsheet directly in BankIt):
Before uploading the file in BankIt, you should also check for any discrepancies in your data (especially the collection_date) that the spreadsheet software might have automatically introduced.
The Features Step/Annotation with the Five-Column Feature Table
The Features step is an essential step in submitting your organelle genome(s). You are required to provide genes, coding regions (CDS), and other feature annotations on your genomes. Submitting your sequences without annotation or inaccurate annotation will delay issuing your accession numbers. GenBank indexers will ask you to resubmit your genomes with added/corrected annotation. It means that you will need to start a new BankIt submission.
BankIt offers two ways of providing features: (1) by completing input forms and (2) by uploading a five-column feature table. Input forms are impractical for annotating organelle genomes with multiple features as you would need to work through the forms for each individual feature. Hence, the five-column feature table is currently your only option to provide annotation for organelle genomes.
The format of the feature table is explained in BankIt at the Features step. Our illustrated example shows a small section of the feature table as the submitter of the Chinese short-limbed skink mitochondrion (the MW327509.1 record) would have prepared:

The top panel (blue background) shows how to format the table using spreadsheet software. Just like with the source table, you need to save your final feature table as a plain-text tab-delimited file before you can import it in BankIt. If you are working directly in a text editor, you need to move from one column to another with a single stroke on the tab key on your computer keyboard.
The total number of columns in the feature table is five. For each feature, locations are provided in columns 1 and 2. For a feature on the complement strand, invert the locations (the larger one of the two should be in column 1). The feature key is in column 3. Qualifiers and their values that follow are offset in columns 4 and 5.
The bottom panel (yellow background) in the image reflects the information in the FEATURES section of the processed GenBank record. The processed record has additional qualifiers that were not in the original feature table. BankIt will use codon_start 1 (reading frame 1) as default if none is provided in the feature table.
You should not be providing protein_ids in a feature table as these will be uniquely assigned for each translated CDS on your genome at the time your record is processed.
The feature file that you import in BankIt should contain the features of all the sequences that you imported at the Nucleotide step. For example, there will be three sets of feature annotations in the file for the three Physalis chloroplast genomes. Each starts with a feature definition that contains the ">" symbol and the word "Feature". The word "Feature" is followed by a space and then by the sequence ID that matches in the corresponding FASTA sequence:

You should always provide the feature locations as they are on the genome that you are submitting. You are required to determine the correct feature locations.
Your annotation software may offer the five-column feature table as one of the annotation outputs. However, as we detail in the troubleshooting section, you need to review the annotation and the table format to meet GenBank (INSDC) standards.
You can also use a feature table from a GenBank record that represents a similar genome to the one that you are submitting as your template. The troubleshooting section provides steps for obtaining a template from the NCBI web and a tip for removing unwanted information from the table.
Once you have prepared the table, upload it in BankIt at the Features step, by selecting the following button:
- Add features by uploading five column feature table file
Uploading the file is a two-step process. First use the Choose File button, then the Upload file button. Upon uploading the table, BankIt will translate the coding regions based on their annotation (locations). If you are working with a known organism, BankIt will recognize the organism and apply the proper translation table (genetic code) for translation. However, if the organism is not yet in the database (unknown), you need to provide the genetic code for each CDS in the table by introducing the transl_table qualifier as we show in our illustrated examples above.
BankIt will also validate your submission. Address any error reports, such as internal stop codons in protein translation. Use other approaches as listed in the troubleshooting section to assure accuracy of your annotation.
Once you complete your submission, GenBank indexers will communicate with you to address any remaining issues and assign accession numbers for your records. Your records will require manual processing before they are publicly released.
Do not send a new submission if you cannot find your records in GenBank. Please write gb-admin@ncbi.nlm.nih.gov and inquire about the status.
Troubleshooting Annotation/Feature Table Format
Annotation programs and copying features from a similar genome is a good way to begin annotating a new genome. However, you need to review the resulting annotations for accuracy and completeness. For example, annotation programs may put on a partial coding region because it cannot determine the complete coding region. Or an essential gene may be annotated as a misc_feature because there is an internal stop codon in translation.
If using an annotation program:
-
Determine that the complete coding region has been annotated. Use other tools such as BLAST to evaluate the accuracy of the predicted starts and stops of the coding regions. If several features are partial this indicates that the annotation program was unable to determine the correct endpoints.
-
Review the product names. The following are examples of product names that you should modify/change:
a. beginning with 'TPA:'
b. containing the phrase 'No product string in profile'
c. containing the word 'partial' or a bracketed term
-
Review the gene features to assure that they have corresponding CDS/tRNA/rRNA features. A gene feature should not be listed or linked with a misc_feature. Lacking CDS features for protein-coding genes usually indicate the annotation software cannot predict a valid conceptual translation due to presence of internal stop codons or missing the start and/or stop codon.
-
You should check your sequence/assembly quality if the annotation tools are failing with CDS annotation of essential genes. You can find guidance on using BLAST for checking sequence quality in a series of knowledgebase articles.
-
Annotation tools may introduce unwanted annotation lines/features that do not meet INSDC standards. Remove the lines/features that contain:
a. misc_features with the annotation program name.
b. the 'label' term.
c. systematic locus tags such as 'gene 1'.
d. the standard_name qualifier
e. BLAT output
If copying features from a similar genome:
-
Exclude (remove): locus_tags (these must be unique for each genome if included), gene_xref, GeneID, and protein_ID lines from the table.
-
Check that the annotation is reasonable. For example, a variation feature on one genome may not apply to your genome.
Downloading and adjusting feature table template:
You might have identified a GenBank record that can serve as an annotation template. Here we use the MT161478.1 record as an example. To download its feature table:
-
Click the Send to link top right above the record.
-
Select: Complete record File Feature table
-
Click the Create File button
The downloaded file will contain protein_id qualifiers (lines) that you need to remove.
You can use various text editors functions to remove repetitive unwanted lines. For example, open the downloaded file with the Notepad++ editor. To remove the lines:
-
Select: Search Mark...
-
Check the Bookmark line check box in the Mark dialogue.
-
Enter: protein_id as your search term in the Find what box of the Mark dialogue.
-
Click the Mark All button.
-
Close the Mark dialogue.
-
Select: Search Bookmark Remove Bookmarked lines
Annotation Checks for individual organism groups:
Here are some of the basic annotation checks that you need to meet for your organism groups:
I. Metazoan Mitochondria
- Each genome should have 13 CDS features, 22 tRNAs and 2 rRNAs.
There are known exceptions such as Cnidaria and Porifera. If your annotation is missing one of the expected features, provide an explanation. You can add a message for the indexers on the last BankIt page, Review and Correct.
-
The CDS feature rarely overlaps the tRNA feature by more than a few nucleotides. These coding regions use the mitochondrial TAA stop.
-
Use protein BLAST to evaluate the length and similarity of the predicted proteins against those from related organisms.
-
With few exceptions (for example mollusks), the features are on both strands. Check the strandedness of the tRNAS and rRNAs.
II. Fungi
- Intronic ORFs should be located within an intron.
III. Embryophyta Plants
-
Rps12 is a trans-spliced gene consisting of 2-3 exons. This gene is not RNA edited.
-
Check the size of the tRNAs. tRNAs can consist of two exons in some cases.
-
Commonly RNA edited genes are ndhD, psbL, rpl2.
-
The ribosomal RNAs that should be annotated: 4.5S, 5S, 16S and 23S ribosomal RNA.
For more help, contact the NCBI helpdesk at: info@ncbi.nlm.nih.gov .
If you need to update already accessioned records, follow the instructions on the GenBank Update page.