

Prokaryotic RefSeq Genome Re-annotation Project
Related documentation
What we are doing?
Prokaryotic RefSeq genomes are being re-annotated using NCBI's annotation pipeline. The scope of this project includes all new WGS and complete genomes and all pre-existing RefSeq WGS and complete genomes with the exception of a small number of identified highest quality reference genomes which have been extensively curated, community-vetted and/or have a large amount of experimental confirmation. Coincident with this update, we are converting prokaryotic RefSeq genomes to a new non-redundant protein data model that was first announced via the NCBI NEWS site and an accompanying longer announcement. Where an important reference genome exists, such as Escherichia coli K12 substr. MG1655, it will continue to be annotated with species and strain-specific RefSeq proteins (with NP_ or YP_ accession prefix) which in turn cross-reference the non-redundant RefSeq protein dataset in the CONTIG line that appears above the protein sequence in the GenPept display format (e.g., NP_414543). Identical proteins in other non-reference genome strains will only be represented by the non-redundant protein accession number (WP_).
The rapidly growing redundancy in the prokaryotic database due to an increase in sequence submissions has caused us to change the scope definition for Gene. The scope of Gene, for prokaryotes, was changed from all complete genomes, to only those genomes that are reference genomes for which there are at least 10 sequenced genomes for the species or clade. In other words, Gene will now include only the highest quality and most supported subset of RefSeq prokaryotic genomes.
We also refined our quality criteria and data validation for prokaryotic genomes. We realize that it is important to provide genome annotation for the multiplicity of bacterial genomes; however, we do not want to release annotation results that may have significant quality issues. We may continue to refine our quality criteria in the coming year which may result in an additional changes. Prokaryotic RefSeq genomes that we feel are not currently of sufficient quality to provide as a RefSeq record(RefSeq genome quality criteria) will be suppressed. Some of these may be added back in the future if the source GenBank genome assembly is updated or following additional improvements to NCBI's prokaryotic genome annotation pipeline.
Why are we doing this?
With the advent of high-throughput sequencing, the generation and use of genome sequences is evolving. One change is the sequencing of very large numbers of nearly identical bacterial genomes to analyze food borne pathogens or infection outbreaks. In these cases, annotating the genes on these genome is important and useful for identifying the small number of places where mutations may affect function, but most of the genes annotated on these new genomes encode proteins that are identical to proteins already in the RefSeq dataset.
This health-related use case makes it critically important to provide genome annotation that has been generated using a consistent method. Previously, RefSeq bacterial genomes represented a mix of submitted and NCBI-generated annotation that was provided using different methods reflecting different states of technology development over time. Therefore, in order to continue to provide a prokaryotic dataset that is of highest utility for disease, pathogen, and other comparative analysis needs, NCBI decided to re-annotate all RefSeq prokaryotic genomes using an improved genome annotation pipeline.
In the past, differences in annotation methods resulted in different protein names and lengths from identical DNA sequences submitted from the same species. These problems will be avoided with the re-annotation project, and the same DNA sequence from the same species will now be annotated with exactly the same protein sequence and name in RefSeq.
How are we doing this?
In order to manage the flood of identical proteins resulting from the decision to provide genome annotation for this volume of sequenced genomes, we introduced a new data model for prokaryotic RefSeq genomes, and a new protein data type in the RefSeq collection which is signified by a "WP_" accession prefix. Previously, each annotated CDS on each RefSeq prokaryotic genome was tracked with a distinct RefSeq protein accession number. This practice led to redundancies in the protein database, since the identical protein was represented by multiple accessions as a result of extensive sequencing of bacterial genomes. To avoid such redundancies in the database, NCBI has adopted the use of non-redundant WP proteins for RefSeq prokaryotic genomes that are annotated using NCBI pipelines. If the identical protein sequence (exactly the same protein sequence and length) appears on more than one RefSeq genome, NCBI simply re-uses the existing WP accession number instead of creating a new accession for each new occurrence and genome. For conserved proteins the same WP accession may appear on thousands of genomes. This is a first step toward dealing with a world when genomes are sequenced just for assays, rather than to discover novel proteins. We appreciate that this is new and a major change for RefSeq prokaryotic genomes, and that there are some issues still to be worked out, but we felt we needed to start making this change as the number of disease-outbreak and other isolate sequencing continues to increase rapidly.
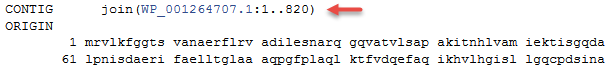
For example, the RefSeq non-redundant 50S ribosomal protein L11, WP_003156430.1, is annotated on over 2000 RefSeq bacterial genomes. The CDS feature annotated on those genomes cross-references this same WP protein accession number, indicated by a red arrow. Examples include:
1) 50S ribosomal protein L11 from Bacillus sp. JS:

2) 50S ribosomal protein L11 from Bacillus subtilis subsp. spizizenii strain NRS 231:

Advantages
Consistent genome annotation
This approach enables direct comparison of RefSeq prokaryotic genome annotation across genomes because annotation is generated using a consistent method. The following figure provides an example of this. To generate this figure, the displayed RefSeq and GenBank genomes were each aligned to subject NC_021200.1, the pair-wise alignments were added to NCBI's graphical sequence display tool (using the Configure tool and selecting the option to add local data, add BLAST RID), and the display was configured to hide the set of primary alignments (leaving only the cleaned and merged alignments) and to display the annotated features (using the Configure tool, select the 'cleaned alignment' row and check the box to project features). The display was then zoomed into the range 2,123 to 2,125 kb. Note that the three RefSeq genomes shown have consistently annotated a gene, between the two vertical blue bars, for phosphoribosyl transferase (229 amino acids) but the annotation differs in the three GenBank genomes both in terms of the protein length and name. For two of the RefSeq genomes, the annotated protein is identical and cross-references a single non-redundant protein (WP_003872216.1), and the third RefSeq genome differs in sequence but not length and cross-references a different non-redundant RefSeq protein (WP_033715495.1). Also note that the GenBank genome AE016958.1 is missing two genes (blue circle) in the displayed region, and the GenBank genome CP005829.1 is missing one gene (not circled).

Reduced protein redundancy
The use of non-redundant RefSeq accessions significantly reduces redundancy in the prokaryotic proteome represented in RefSeq. For example, the non-redundant protein WP_003872216.1 is annotated on 26 RefSeq genomes of Micobacterium avium subsp. paratuberculosis; this is a 26 fold reduction in redundancy for this one protein sequence in the RefSeq collection as only one RefSeq protein accession is annotated on these 26 genomes, instead of creating 26 accessioned records - one for each genome. The reduction can be even greater for other highly conserved proteins that are annotated on multiple hundred, or even over a thousand, RefSeq genomes. For example, WP_000289090.1 is annotated on over 500 genomes of Escherichia and Shigella. Each of these RefSeq genomes has an annotated CDS feature that cross-references this single protein accession.
Improved name management
As protein names are updated on the non-redundant RefSeq protein records, the improved protein name will automatically become available on the annotated genome record which cross-references the non-redundant dataset. We are continuing to work on protein name improvements by leveraging information from: a) curated trusted protein datasets for families; b) curated Swiss-Prot names; c) community curated names on reference genomes; d) Hidden Markov Models (HMMs); e) conserved domains; and f) protein clustering approaches.
Easy navigation
Protein records now include an "Identical Proteins" link, which provides convenient access to all 100% identical proteins as well as their annotated locations. For example, all genomes containing a protein identical to WP_013315922.1 can be reviewed through the Identical Proteins report, allowing quick review of species and strain diversity for this protein as well as the option to view the corresponding genomic annotations.

Description of data changes
-
Re-annotated genomes: Over 32,000 prokaryotic RefSeq genomes have now been annotated using NCBI s improved prokaryotic genome annotation pipeline. At the same time, the approach used to annotate CDS features and represent RefSeq proteins was change to use the non-redundant protein data model.
-
Finished transition to new data model: The new data model is now in use for both WGS and Complete bacterial and archaeal genomes.
- This data model was first introduced in mid-2013 and has been in place since then for NCBI annotations of whole genome shotgun sequences.
- As planned, we have now finished transitioning all WGS and complete prokaryotic genomes to this new model with the exception of a small number of selected reference genomes.
- Now, genomes are annotated with non-redundant proteins (WP_ accessions).
-
Reference genomes: For each defined species with assemblies included in RefSeq, one assembly is designated as 'reference'. Reference genomes are a compact, normalized, and taxonomically diverse view of the RefSeq collection that can be used for the taxonomic identification and characterization of novel sequences. Reference genomes are selected based on considerations that include genome assembly and annotation quality. Selection criteria are described here. With the exception of a few manually selected genome assemblies, reference genomes are annotated with non-redundant RefSeq protein accessions (WP_ accessions). A small subset of reference genomes are selected based on a long history of collaboration and wide recognition as a community standard, such as the reference genome of Escherichia coli str. K-12 substr. MG1655, or based on medical importance, sequence and annotation quality, and the availability of experimental support. This subset is not part of the reannotation project and will continue to have the NP_ or YP_ accessions and annotation provided by the submitters. The Reference genome dataset supports those who want a minimal prokaryotic dataset of the highest quality. The reference genomes can be found in the Datasets database or on our FTP site.
-
Suppressed genomes: RefSeq bacterial genomes that do not pass assembly or annotation quality validation have been suppressed(RefSeq genome quality criteria). An FTP report file listing the most recent suppressions can be found on the FTP site.
-
Suppressed proteins: Approximately 7 million prokaryotic NP_ and YP_ accessioned proteins have been suppressed as a result of both suppressing RefSeq genomes with quality issues and due to the large-scale re-annotation project which transitioned bacterial genomes to the new non-redundant protein data model. The new data model provides CDS annotation that cross-references non-redundant RefSeq proteins. This data model applies to all prokaryotic genomes with the exception of the select subset of reference genomes mentioned above.
-
Changes in the Gene resource: The scope definition for annotating prokaryotic genomes with NCBI GeneIDs was changed resulting in the suppression of a large number of entries. Gene continues to provide current entries for all prokaryotic reference genomes. For prokaryotes, Gene includes the best supported set of reference genomes for which there are >= 10 sequenced genomes for the clade. Suppressed Gene entries can still be accessed and we are supplementing the current set of suppressed records with information to facilitate navigating to the replacement non-redundant RefSeq protein. The embedded graphical display will continue to show annotation of the genomic coordinates that the Gene entry represents. If that RefSeq genome was re-annotated, then the display in Gene will automatically show the updated annotation for the accession.version:from-to coordinates associated with the Gene record. Thus, while the Gene entry may have initially displayed a CDS annotation associated with a YP_ accession number (still reported in the Reference Sequences section of the record), it may now display a CDS annotation associated with a non-redundant WP_ accession number. These records will not be subject to any further update.
-
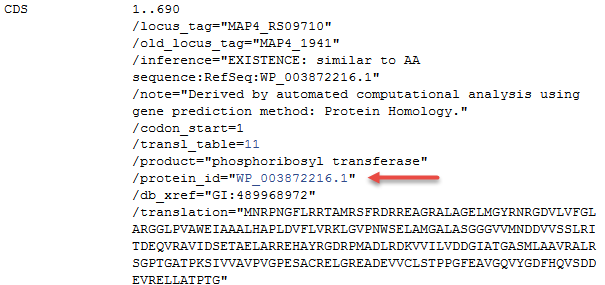
Locus_tag changes: New locus_tags were assigned as genomes were re-annotated using the format of <original locus_tag prefix>_RS<digits>, for example "MAP4_RS09710". The locus_tag that was previously annotated on this genome is reported in the 'old_locus_tag' qualifier when the equivalent CDS was re-annotated on the record. In some cases a new, or substantially modified, CDS was annotated on the genome in which case there is no corresponding previous locus_tag. In order to continue to provide locus_tag annotation comprehensively on these records we had to provide it in a new format.

What we are doing to help you transition to the new data
We are working to provide information to facilitate the transition to the new annotation data using several approaches:
-
Data mapping and other report files: We provided supplemental report files on the RefSeq FTP site, with RefSeq release 70, which can be used to identify removed records, and to map removed records to the non-redundant protein accessions. These files are available in the release archive directory at: ftp://ftp.ncbi.nlm.nih.gov/refseq/release/release-catalog/archive/. Two files are provided that report: a) mapped protein accessions and locus_tags, for the set of NP_ or YP_ accessioned proteins that were suppressed and replaced by a nonredundant WP_ accessioned protein; and b) a set of assemblies that were suppressed between March and May 2015 due to changes in the RefSeq quality criteria.
-
Suppressed proteins: One or more files will be provided in the RefSeq FTP site that map suppressed NP_ and YP_ protein accessions to non-redundant WP_ accessions which are identical in sequence and length to the suppressed protein record. Because these updates have occurred over time we are planning to provide a comprehensive report of all previously suppressed NP_ and YP_ proteins that can be mapped to a non-redundant WP_ accession. Note that this is mapping at the level of protein sequence identity, not at the level of genome annotation, and this report may include some suppressed accessions that were removed a number of years prior to development of our re-annotation plans. In addition, the RefSeq release FTP site includes a report of accessions that have been removed since the previous release. This file can be parsed to identify all removed bacterial records. An example command line that identifies removed bacterial protein accessions is:
- >zcat release69.removed-records.gz |grep bacteria|grep P\_|more -
Locus_tags: We will also provide mapping data between old and new locus_tags for those annotated genes and CDSs which are identical or highly similar to the previous version of the annotated genome. This data is also available directly within the re-annotated RefSeq genomes as the previous locus_tag has been provided in the "/old_locus_tag:" qualifier.
-
Suppressed genomes: We will provide a report of RefSeq bacterial genomes that were removed due to concerns about either the assembly or the annotation. In some cases, the quality issue identified requires an update to NCBI's prokaryotic genome annotation pipeline. Once the pipeline software has been updated, then the affected genomes will be updated and made publicly available again. In other cases, where the issue is about the assembled sequence quality, the RefSeq genome would only become publicly available again if an improved quality update is submitted to GenBank, ENA, or DDBJ.
-
Suppressed Gene entries: The Gene FTP site provides a file (gene_history.gz) reporting all removed records for all taxa. This file can be parsed to find removed entries for a given date range or NCBI taxonomy ID. An example command line argument is: - >zcat gene_history.gz | grep 2015 | grep ^470 | more
-
Navigation from non-redundant proteins to genomic locations and other resources: Non-redundant protein records do not contain markup of their individual genomic locations or locus_tags. They may also be identified as MULTISPECIES (for example WP_004299620.1), meaning the protein is found in more than one species. Several links are available to help facilitate using these records:
- The "Identical Proteins" link at the top of the record can be used to view a table of all genomic locations for the protein accession, as well as other 100% identical proteins. This table provides convenient links to each annotated genomic record in GenBank format. See the above image.
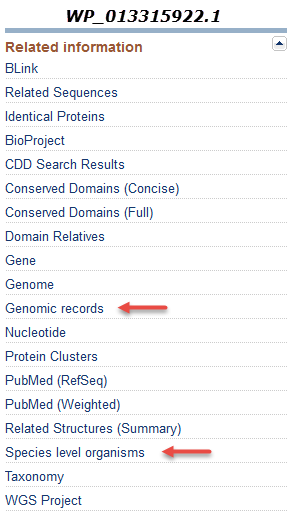
- The "Genomic records" link under "Related Information" (shown below) can be used to navigate to the set of nucleotide records on which the protein is annotated.
-
The "Species level organisms" link under "Related Information" can be used to find the set of species on which this specific non-redundant protein record has been annotated.
-
Navigation from suppressed protein records to replacement non-redundant records: When accessing a suppressed NP_ or YP_ protein sequence in NCBI's Protein resource, an informational message will appear on suppressed sequences that are identical to a non-redundant RefSeq protein record (see below). The message will include links to the replacement WP_ accession, the Identical Protein display, and this documentation. Initially this message will be provided when viewing the record (GenBank format) and for suppressed records for which there is an identical non-redundant sequence record. We are working to provide a similar message on suppressed records for which there is a similar but not identical non-redundant RefSeq record, and to expose this or similar text in other contexts (Summary view as one example). The additional links may not be available in all places due to technical details of how different display options are managed.

-
Navigation from removed Gene entries to replacement non-redundant records: Gene entries that were suppressed due to the revised scope definition continue to be publicly available; however, those GeneIDs are no longer annotated in the corresponding RefSeq sequence record and are not subject to further updates over time. Suppressed Gene records will be modified to provide access to the GenPept Identical Protein report page that can be used to determine valid genomic locations and organism information for the appropriate non-redundant protein record. Please be aware that the graphical display will automatically show the current annotation for the genomic location that the Gene record represented at the time it was suppressed.
-
BLAST: We are evaluating technical aspects of some modifications to BLAST results to improve access to species, strain, and genome annotation information for BLAST hits to non-redundant WP_ accessions. This improvement will be added as soon as possible and some information may be easier to add before other content.
-
Documentation and announcements
- We have added extensive documentation for Prokaryotic genomes, non-redundant proteins, and the re-annotation project to the RefSeq web site. We anticipate that these documents will continue to be expanded upon in the coming weeks and months.
- Announcements of changes to RefSeq policy and data content are distributed to subscribers to the refseq-announce email list. In addition, announcements of significant changes are distributed in the NCBI NEWS site, NCBI Staff Twitter account and more.