Summary
Clinical characteristics.
Hepatic veno-occlusive disease with immunodeficiency (VODI) is characterized by (1) combined immunodeficiency and (2) terminal hepatic lobular vascular occlusion and hepatic fibrosis manifesting as hepatomegaly and/or hepatic failure. Onset is usually before age six months. The immunodeficiency comprises severe hypogammaglobulinemia, clinical evidence of T-cell immunodeficiency with normal numbers of circulating T and B cells, absent lymph node germinal centers, and absent tissue plasma cells. Bacterial and opportunistic infections including Pneumocystis jirovecii infection, mucocutaneous candidiasis, and enteroviral or cytomegalovirus infections occur. In the past the prognosis for affected individuals was poor, with 100% mortality in the first year of life if unrecognized and untreated with intravenous or subcutaneous immunoglobulin (IVIG/SCIG) and Pneumocystis jirovecii prophylaxis. However, with early recognition and treatment, including the more recent use of defibrotide, there is a marked improvement in prognosis. Early hematopoietic stem cell transplantation (HSCT) using non-hepatoxic drugs in conditioning and prophylactic defibrotide is potentially curative.
Diagnosis/testing.
The diagnosis of VODI is established in a proband who meets clinical diagnostic criteria or by identification of biallelic pathogenic variants in SP110 on molecular genetic testing.
Management.
Targeted therapies: IVIG/SCIG; defibrotide for acute hepatic disease; HSCT with non-hepatotoxic conditioning therapy, preferably with defibrotide prophylaxis.
Supportive care: Pneumocystis jirovecii prophylaxis; prompt treatment of infections with antibacterials, antivirals, or antifungals as indicated; standard treatment for complications of liver disease; consider liver transplantation, although rate of complications may be high.
Surveillance: Assess growth every three to six months; measurement of immunoglobulin concentrations every three to six months; bronchoalveolar lavage to diagnose Pneumocystis jirovecii infection; viral and bacterial cultures and PCR as needed; serum aminotransferases, bilirubin, albumin, complete blood count, and platelet count every three to six months; pulmonary function studies annually once able to perform reliably; cerebrospinal imaging to identify leukodystrophy or other central nervous system pathology when clinically indicated.
Agents/circumstances to avoid: Agents known to predispose to hepatic veno-occlusive disease including cyclophosphamide and Senecio alkaloids / bush teas.
Evaluation of relatives at risk: If both pathogenic variants in the family are known, it is appropriate to evaluate via molecular genetic testing sibs of a proband who are younger than age 12 months in order to identify those who would benefit from initiation of IVIG or SCIG treatment, Pneumocystis jirovecii prophylaxis, and consideration of preemptive HSCT.
Genetic counseling.
VODI is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for an SP110 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the SP110 pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives and prenatal/preimplantation genetic testing for VODI are possible.
Diagnosis
Clinical diagnostic criteria for hepatic veno-occlusive disease with immunodeficiency (VODI) have been established [Roscioli et al 2006, Ganaiem et al 2013].
Suggestive Findings
VODI should be suspected in individuals with the following clinical, laboratory, imaging, and histopathologic findings and family history.
Clinical findings
- Clinical evidence of immunodeficiency with bacterial and opportunistic infections including Pneumocystis jirovecii infection, mucocutaneous candidiasis, and enteroviral or cytomegalovirus infections
- Hepatomegaly or evidence of hepatic failure, not explained by other factors, in the affected individual or a first-degree relativeNote: Hepatic veno-occlusive disease (hVOD; also known as sinusoidal obstruction syndrome) is usually present and pathognomonic but may not be found, or may have resolved.
- Onset usually before age six months
Laboratory findings
- Low serum concentrations of immunoglobulin A (IgA), IgM, and IgGNote: Immunoglobulin levels are age specific and laboratory specific and thus should be compared against appropriate local reference ranges.
- Normal lymphocyte numbers and CD4 and CD8 percentages
- Normal lymphocyte proliferative responses to mitogens
Imaging findings
- Hepatic ultrasonography. Features consistent with hVOD may include hepatosplenomegaly, gallbladder wall thickening, increased portal vein diameter, reduced hepatic vein diameter, ascites, and recanalization of the ligamentum teres.
- Doppler ultrasound examination. Features consistent with hVOD may include reduced portal venous flow, flow in the paraumbilical vein, and increased resistance in the hepatic artery.
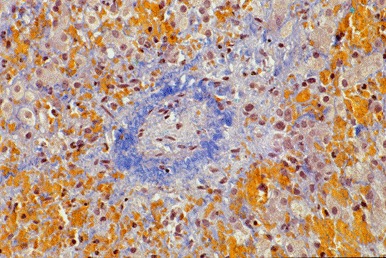
Histopathologic findings. Features consistent with hVOD may include fibrous concentric narrowing of zone 3 terminal hepatic venules, centrilobular hepatocyte necrosis, and sinusoidal congestion (see Figure 1). Note: If hepatic biopsy is contraindicated, hepatic ultrasonography and Doppler ultrasonography may provide supportive evidence of hVOD.

Figure 1.
Hepatic biopsy showing vascular obliteration, perivenular fibrosis, zone 3 fibrosis, and hepatocyte dropout from a girl who presented at age five months with hepatomegaly and ascites (Picro-Mallory stain, 100x)
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The clinical diagnosis of VODI can be established in a proband based on clinical diagnostic criteria, or the molecular diagnosis can be established in a proband using molecular genetic testing.
Clinical Diagnosis
The clinical diagnosis of VODI can be established in a proband with ALL of the following clinical diagnostic criteria [Roscioli et al 2006, Ganaiem et al 2013]:
- Hypogammaglobulinemia with clinical evidence of T-cell immunodeficiency with bacterial and opportunistic infections including Pneumocystis jirovecii infection, mucocutaneous candidiasis, and enteroviral or cytomegalovirus infections
- Hepatomegaly or evidence of hepatic failure, not explained by other factors, in the affected individual or a first-degree relative
- Onset prior to age 12 months
- Family history consistent with autosomal recessive inheritance
Molecular Diagnosis
The molecular diagnosis can be established in a proband with suggestive findings and biallelic pathogenic (or likely pathogenic) variants in SP110 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of biallelic SP110 variants of uncertain significance (or of one known SP110 pathogenic variant and one SP110 variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing). Gene-targeted testing requires that the clinician determine which gene(s) are likely involved (see Option 1), whereas comprehensive genomic testing does not (see Option 2).
Option 1
Single-gene testing. Sequence analysis of SP110 is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
Note: Targeted analysis for the c.642delC pathogenic variant can be performed first in individuals of Lebanese ancestry (see Table 7).
A multigene panel that includes SP110 and other genes of interest (see Differential Diagnosis) may be considered to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the diagnosis of VODI has not been considered because an individual has atypical features, comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible. To date, the majority of SP110 pathogenic variants reported (e.g., missense, nonsense) are within the coding region and are likely to be identified on exome sequencing.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Hepatic Veno-Occlusive Disease with Immunodeficiency
Clinical Characteristics
Clinical Description
Hepatic veno-occlusive disease with immunodeficiency (VODI) is a primary immunodeficiency associated with terminal hepatic lobular vascular occlusion and hepatic lobule zone 3 fibrosis. Infants typically present prior to age six months with tachypnea, poor weight gain, interstitial pneumonitis, jaundice, ascites, hypogammaglobulinemia, and thrombocytopenia. Some individuals develop neurologic sequelae (cerebral infarction, leukodystrophy). The following description of the phenotypic features associated with this condition is based on published case series [Roscioli et al 2006, Cliffe et al 2012, Wang et al 2012, Ganaiem et al 2013, Marquardsen et al 2017] supported by additional reports [M Wong, personal communication].
Table 2.
Hepatic Veno-Occlusive Disease with Immunodeficiency: Frequency of Select Features
Immunodeficiency is characterized by severe hypogammaglobulinemia, clinical evidence of T-cell immunodeficiency with normal numbers of circulating T and B cells, absent lymph node germinal centers, and absent tissue plasma cells [Roscioli et al 2006, Cliffe et al 2012, Wang et al 2012, Ganaiem et al 2013, Marquardsen et al 2017]. Bacterial and opportunistic infections including Pneumocystis jirovecii pneumonitis (PJP), mucocutaneous candidiasis, and enteroviral or cytomegalovirus infections occur. Presentation to medical care at about age two to six months is typically acute, with respiratory symptoms (often from PJP) and abdominal distention, followed by rapid clinical deterioration. There is, however, often a preexisting history of feeding difficulties and diarrhea, with associated weight loss and poor growth in the previous month(s). Oral candidiasis (thrush) may be present. Early recognition and treatment of PJP with high-dose cotrimoxazole and steroids and cytomegalovirus with ganciclovir are essential. Other viruses may persist due to impaired clearance, causing ongoing complications such as chronic diarrhea.
Hepatic veno-occlusive disease (hVOD). Children with VODI present ab initio either with hepatomegaly (83% with a history of preceding infection) or hepatic failure (53% with a history of preceding infection). Asymptomatic increase in liver transaminases, thrombocytopenia, and anemia is commonly detected in both scenarios. Affected infants with hepatic failure typically present with jaundice, ascites, and hepatosplenomegaly, with subsequent rapid clinical deterioration and evidence of functional deficits such as bleeding from coagulopathy and encephalopathy, sometimes necessitating liver transplantation. The results of liver transplantation in this setting are poor, with few children surviving [Wozniak et al 2011, Cliffe et al 2012]. The mortality rate of hVOD overall is high. Even if successful, liver transplant does not cure the immunodeficiency.
In individuals with less severe disease, treatment of the precipitating infection, commencement of long-term immunoglobulin replacement therapy (intravenous immunoglobulin [IVIG] or subcutaneous immunoglobulin [SCIG]), continuation of prophylactic cotrimoxazole, and other supportive measures may result in stabilization of liver function, and the liver gradually heals by nodular regeneration. Associated hepatosplenomegaly, anemia, and thrombocytopenia also resolve. Affected individuals may continue to have compromised liver function. Although maintenance immunoglobulin replacement therapy and cotrimoxazole prophylaxis significantly reduces the risk, recurrence of hVOD can occur, especially precipitated by infection or other metabolic stress.
The hepatic injury in VODI is identical clinically and histologically to hVOD (also known as sinusoidal obstruction syndrome) following high-dose myeloablative conditioning therapy in hematopoietic stem cell transplantation (HSCT) [Roscioli et al 2006]. Early initiation of defibrotide, an anti-inflammatory, antithrombotic drug that restores endothelial thrombotic-fibrinolytic balance, has been demonstrated to improve survival in post-HSCT-associated sinusoidal obstruction syndrome [Richardson et al 2017], for which it is now approved therapy [Carreras 2015]. The duration of treatment is usually 21 days. Similarly, early initiation of defibrotide has been associated with rapid response in VODI-associated liver disease [M Wong, personal observation]. In addition, based on clinical trial data suggesting a protective effect in high-risk individuals undergoing HSCT [Corbacioglu et al 2012], prophylactic defibrotide is increasingly being used in the peri-transplant period in children with VODI [M Wong, personal observation].
Neurologic manifestations. Overall, 30% of children with VODI had neurologic involvement; none of the individuals were reported to have veno-occlusive disease of the brain [Roscioli et al 2006, Cliffe et al 2012]. Four individuals had extensive cerebral necrosis on postmortem examination. A striking finding is the presence of cerebrospinal leukodystrophy in three individuals (20%) with VODI. One affected female remained well on IVIG and cotrimoxazole prophylaxis until age 18 years, when she suddenly developed paraparesis and urinary retention associated with cerebral lesions. No infective cause or evidence of veno-occlusive disease was found on extensive investigation. There was partial response to high-dose IVIG and steroid therapy, suggesting an inflammatory etiology [M Wong, personal observation].
It is unclear whether other neurologic features in single reports are complications of VODI or coincidental findings. These include syndrome of inappropriate antidiuretic hormone secretion (SIADH) during the presenting illness, microcephaly, nonspecific calcified central nervous system (CNS) lesions on imaging, developmental delay, and attention-deficit/hyperactivity disorder (ADHD).
Prognosis. VODI is associated with 100% mortality in the first year of life if unrecognized and untreated with IVIG or SCIG replacement and Pneumocystis jirovecii prophylaxis, and 90% mortality overall by the mid-teenage years despite initial response to immunoglobulin replacement therapy, cotrimoxazole prophylaxis, and early treatment of infections [Roscioli et al 2006, Cliffe et al 2012]. There may be an ongoing risk for neurologic inflammatory complications. The oldest known individual with VODI is now age 30 years. She remains paraplegic after an unexplained episode of CNS inflammation at age 18 years, but is otherwise stable on IVIG and cotrimoxazole prophylaxis.
Milder phenotypes associated with missense pathogenic variants have been reported [Wang et al 2012, Delmonte et al 2020]. One of these children presented older than age one year with bacterial rather than opportunistic infections and developed liver disease with nodular regenerative hyperplasia but no hVOD while on IVIG [Delmonte et al 2020]. The other presented at age five months with PJP and hVOD but recovered rapidly after treatment of PJP and the first dose of IVIG and was well at the time of reporting (age 3 years) on IVIG and cotrimoxazole prophylaxis [Wang et al 2012]. This child, however, had a sib who died at age three months from hepatic failure of undetermined etiology, presumably VODI. Both individuals had normal SP110 mRNA but impaired/absent protein expression.
The poor long-term prognosis and the fact that SP110 is primarily expressed in leukocytes has led to consideration of HSCT as a potential cure, preventing further recurrences of hVOD and other manifestations of immune dysregulation such as CMS inflammation as well as removing the need for immunoglobulin replacement therapy and cotrimoxazole prophylaxis.
There are limited clinical reports of outcomes following HSCT for VODI. The first published report of HSCT for VODI described five individuals from one extended family (out of a total of 14 affected family members) who had matched related donor transplants [Ganaiem et al 2013]. All five individuals received conditioning with fludarabine and serotherapy (4 received antithymocyte globulin, 1 received alemtuzumab) and an alkylating agent (cyclophosphamide, busulfan, or treosulfan). The two individuals who also received thiotepa, another alkylating agent, died following engraftment with hVOD and multiorgan failure. Two of the other three were alive and well and the third was alive with epilepsy and severe ADHD at the time of reporting, between 11 months and 6.5 years post transplant. The results suggest that the additional thiotepa increased the risk of hVOD.
One child presented at age four months with probable herpetic hepatitis that initially responded clinically to acyclovir, but then developed florid hVOD following a transfusion. She responded well to defibrotide, IVIG, and prophylactic cotrimoxazole. She was neurologically normal, but asymptomatic bilateral subdural hematomas were found on routine pre-transplant MRI despite a normal head ultrasound a week prior when coagulation studies and platelets had already normalized. At age six months she underwent an unrelated cord blood transplant with alemtuzumab, fludarabine, treosulfan, and antithymocyte globulin conditioning. Defibrotide was prophylactically continued until two months post transplant. She remained well and neurologically normal at age ten years. Her MRI scan at age two years showed no anatomic abnormality [M Wong, personal obervation].
Another infant presented at age four months with PJP and CMV infections, hVOD, and hypogammaglobinemia. At age five months, after control of pneumonitis and hVOD, he received a haploidentical paternal (CMV positive) HSCT after depletion of α/β TCR and CD19+ cells. He received conditioning with fludarabine, treosulfan, and antithymocyte globulin. Defibrotide was given as hVOD prophylaxis. Two months post transplant he was well and had engrafted without graft-vs-host disease or recurrence of hVOD but with moderate CMV hepatitis. Defibrotide was able to be discontinued at Day 60. Seventy days following HSCT he developed seizures secondary to CMV reactivation with CNS involvement, leading to his death [T Cole, J Smart, & T Soosay Raj, personal communication].
This child's sib was diagnosed soon after birth and underwent HSCT with alemtuzumab, fludarabine, treosulfan, and antithymocyte globulin conditioning and defibrotide prophylaxis at age nine weeks. He was alive and well at last review, age six years.
A further infant, whose brother died in infancy from hemophagocytic lymphohistiocytosis (HLH) and had biallelic SP110 pathogenic variants, presented at age three months with a six-week history of recurrent viral infections, diarrhea, and poor weight gain. She rapidly deteriorated with fever, respiratory distress, oxygen requirement, and hepatomegaly followed by ascites and liver dysfunction. PJP was confirmed and treated with high-dose cotrimoxazole and steroids, while IVIG and defibrotide were commenced with rapid clinical response. She is awaiting transplant with alemtuzumab, fludarabine, treosulfan, and antithymocyte globulin conditioning and defibrotide prophylaxis, likely to occur by age six months.
Genotype-Phenotype Correlations
No significant difference in the clinical manifestations of VODI is observed between individuals with different pathogenic variants. The majority of SP110 pathogenic variants are small deletions or duplications leading to a frame shift with consequent protein truncation. Milder phenotypes may be seen in individuals with SP110 missense variants; however, reports of affected family members dying in infancy with typical VODI presentations precludes genotype-phenotype correlation [Delmonte et al 2020].
Nomenclature
Hepatic veno-occlusive disease alone was known previously as Jamaican bush tea disease due to a dietary and geographic association. This term is now superseded by hepatic veno-occlusive disease (hVOD) or sinusoidal obstruction syndrome (SOS), terms less limiting given the occurrence of hVOD worldwide and it being secondary to other precipitants. The combination of hVOD and a combined immunodeficiency is termed hepatic veno-occlusive disease with immunodeficiency (VODI).
Prevalence
The majority of children reported with VODI are of Lebanese origin due to founder pathogenic variant c.642delC. The prevalence of VODI in the Lebanese population of Sydney, Australia, has been calculated at one in 2,500 [Roscioli et al 2006]. VODI has also been identified in individuals of Lebanese ancestry residing in other regions, as well as individuals of other ancestral origins, with published reports including individuals of Hispanic American [Wang et al 2012], Italian [Cliffe et al 2012], Palestinian [Ganaiem et al 2013], and Pakistani origin [Cliffe et al 2012, Delmonte et al 2020].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline biallelic or heterozygous pathogenic variants in SP110.
Differential Diagnosis
Hepatic veno-occlusive disease (hVOD). The primary differential diagnosis for hVOD alone is environmental alkaloid or sinusoidal cell toxicity. Hepatic veno-occlusive disease has also been reported in association with alcoholic cirrhosis [Kishi et al 1999], ataxia-telangiectasia [Srisirirojanakorn et al 1999], osteopetrosis [Corbacioglu et al 2006] (see CLCN7-Related Osteopetrosis), and hypereosinophilic syndrome (OMIM 607685).
Immunodeficiency. Other combined immunodeficiency disorders and HIV should be considered in the differential diagnosis for the immune phenotype.
To date, there has been no report of SP110 pathogenic variants in individuals described to have hVOD without immunodeficiency.
Management
No clinical practice guidelines for hepatic veno-occlusive disease with immunodeficiency (VODI) have been published. In the absence of published guidelines, the following recommendations are based on the authors' personal experience managing individuals with this disorder.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with VODI, the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Hepatic Veno-Occlusive Disease with Immunodeficiency: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
Targeted Therapies
In GeneReviews, a targeted therapy is one that addresses the specific underlying mechanism of disease causation (regardless of whether the therapy is significantly efficacious for one or more manifestation of the genetic condition); would otherwise not be considered without knowledge of the underlying genetic cause of the condition; or could lead to a cure. —ED
Table 4.
Hepatic Veno-Occlusive Disease with Immunodeficiency: Targeted Therapies
Supportive Care
Supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 5).
Table 5.
Hepatic Veno-Occlusive Disease with Immunodeficiency: Treatment of Manifestations
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 6 are recommended.
Table 6.
Hepatic Veno-Occlusive Disease with Immunodeficiency: Recommended Surveillance
Agents/Circumstances to Avoid
Agents known to predispose to hepatic veno-occlusive disease (e.g., cyclophosphamide and Senecio alkaloids / bush teas) should be avoided.
Evaluation of Relatives at Risk
It is appropriate to evaluate sibs of a proband who are younger than age 12 months in order to identify as early as possible those who would benefit from initiation of intravenous or subcutaneous immunoglobulin treatment, Pneumocystis jirovecii prophylaxis, and consideration of preemptive hematopoietic stem cell transplantation. The majority of children with VODI present before age six months; however, as one child presented at age 11 months, testing should be considered in sibs of a proband who are younger than age 12 months. Evaluations include:
- Molecular genetic testing if the pathogenic variants in the family are known;
- Serum immunoglobulins, complete blood count, and liver function tests at birth and repeated at age six months if the pathogenic variants in the family are not known.
All individuals reported to date with biallelic pathogenic variants in SP110 have exhibited clinical findings within the first year of life; thus, molecular genetic testing of healthy at-risk sibs of a proband who are older than age 12 months is not clinically indicated.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Hepatic veno-occlusive disease with immunodeficiency (VODI) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are presumed to be heterozygous for an SP110 pathogenic variant.
- If a molecular diagnosis has been established in the proband, molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for a SP110 pathogenic variant and to allow reliable recurrence risk assessment.
- If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, it is possible that one of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017]. If the proband appears to have homozygous pathogenic variants (i.e., the same two pathogenic variants), additional possibilities to consider include:
- A single- or multiexon deletion in the proband that was not detected by sequence analysis and that resulted in the artifactual appearance of homozygosity;
- Uniparental isodisomy for the parental chromosome with the pathogenic variant that resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for an SP110 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- All individuals reported to date with biallelic pathogenic variants in SP110 have exhibited clinical findings within the first year of life.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. The offspring of an individual with VODI are obligate heterozygotes (carriers) for a pathogenic variant in SP110.
Other family members of a proband. Each sib of the proband's parents is at a 50% risk of being a carrier of an SP110 pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the SP110 pathogenic variants in the family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk sibs younger than age 12 months for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
- Carrier testing should be considered for the reproductive partners of individuals affected with VODI and individuals known to be carriers of an SP110 pathogenic variant, particularly if consanguinity is likely and/or if both partners are of the same ancestry. The majority of children of Lebanese origin with VODI have the founder pathogenic variant c.642delC [Roscioli et al 2006, Cliffe et al 2012].
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Once the SP110 pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing for VODI are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal and preimplantation genetic testing. While most health care professionals would consider use of prenatal and preimplantation genetic testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- International Patient Organization for Primary Immunodeficiencies (IPOPI)United KingdomPhone: +44 01503 250 668Fax: +44 01503 250 668Email: info@ipopi.org
- Jeffrey Modell Foundation/National Primary Immunodeficiency Resource CenterEmail: info@jmfworld.org
- European Society for Immunodeficiencies (ESID) RegistryEmail: esid-registry@uniklinik-freiburg.de
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Hepatic Veno-Occlusive Disease with Immunodeficiency: Genes and Databases
Table B.
OMIM Entries for Hepatic Veno-Occlusive Disease with Immunodeficiency (View All in OMIM)
Molecular Pathogenesis
SP110 is expressed primarily in leukocytes and spleen; it is induced by interferon gamma and all-trans retinoic acid (ATRA). SP110 encodes Sp110 nuclear body protein and has three major isoforms (isoform A, B, and C). Isoform B is a potent transcriptional corepressor of retinoic acid receptor alpha (RARα), perhaps via competitive exclusion of activators at the receptor [Watashi et al 2003].
Sp110 nuclear body protein is a member of the Sp100/Sp140 promyelocytic leukemia nuclear body (PML NB) protein family. The protein has an Sp100 domain (amino acids 6-159), which is involved in dimerization with other Sp100 family proteins, a nuclear localization signal (amino acids 288-306), and a nuclear hormone interaction domain (LXXLL type), which may act as an ATRA response element. Other domains that are common features of modular proteins involved in chromatin-mediated gene transcription include a SAND domain (amino acids 452-532), a plant homeobox domain (amino acids 537-577), and a bromodomain (amino acids 606-674) [Bloch et al 2000].
Sp110 nuclear body protein associates with the PML NB, a nuclear macromolecular complex, which is deployed to areas of active host or viral DNA replication, transcription, and repair and has been reported to be involved in apoptosis, cell cycle control, and the immune response [Hsu & Kao 2018, Fraschilla & Jeffrey 2020].
Epstein-Barr virus-transformed B cells from an individual with hepatic veno-occlusive disease with immunodeficiency (VODI) and a homozygous inactivating SP110 variant show an absence of nuclear Sp100-specific immunolabeling in a setting of normal numbers of PML nuclear bodies. This finding is consistent with Sp110 nuclear body protein having an important role in the immune response without being essential for PML nuclear body formation [Roscioli et al 2006].
There is no mouse model of Sp110 nuclear body protein deficiency, and the mechanisms leading to clinical disease are yet to be fully elucidated. It is currently unknown whether hepatic veno-occlusive disease (hVOD) is a direct manifestation of SP110 pathogenic variants, related to altered apoptosis in the hepatic sinusoid, or secondary to infection; however, hVOD appears to develop after infections occur [Cliffe et al 2012].
Mechanism of disease causation. Loss of function
Table 7.
SP110 Pathogenic Variants Referenced in This GeneReview
Chapter Notes
Author History
Michael Buckley, MBChB, PhD, FRCPA, FHGSA; Prince of Wales Hospital (2007-2025)
Tony Roscioli, MBBS, FRACP, PhD; Sydney Children's Hospital (2007-2025)
Melanie Wong, MBBS, FRACP, FRCPA, PhD (2007-present)
John B Ziegler, MBBS, FRACP, MD; Sydney Children's Hospital (2007-2025)
Revision History
- 23 January 2025 (sw) Comprehensive update posted live
- 12 January 2017 (sw) Comprehensive update posted live
- 3 July 2013 (me) Comprehensive update posted live
- 15 September 2009 (me) Comprehensive update posted live
- 21 February 2007 (me) Review posted live
- 29 November 2006 (mb) Original submission
References
Literature Cited
- Bloch DB, Nakajima A, Gulick T, Chiche JD, Orth D, de La Monte SM, Bloch KD. Sp110 localizes to the PML-Sp100 nuclear body and may function as a nuclear hormone receptor transcriptional coactivator. Mol Cell Biol. 2000;20:6138–46. [PMC free article: PMC86089] [PubMed: 10913195]
- Carreras E. How I manage sinusoidal obstruction syndrome after haematopoietic cell transplantation. Br J Haematol. 2015;168:481–91. [PubMed: 25401997]
- Cliffe ST, Bloch DB, Suryani S, Kamsteeg EJ, Avery DT, Palendira U, Church JA, Wainstein BK, Trizzino A, Lefranc G, Akatcherian C, Megarbané A, Gilissen C, Moshous D, Reichenbach J, Misbah S, Salzer U, Abinun M, Ong PY, Stepensky P, Ruga E, Ziegler JB, Wong M, Tangye SG, Lindeman R, Buckley MF, Roscioli T. Clinical, molecular, and cellular immunologic findings in patients with SP110-associated veno-occlusive disease with immunodeficiency syndrome. J Allergy Clin Immunol. 2012;130:735–742.e6. [PubMed: 22621957]
- Corbacioglu S, Cesaro S, Faraci M, Valteau-Couanet D, Gruhn B, Rovelli A, Boelens JJ, Hewitt A, Schrum J, Schulz AS, Muller I, Stein J, Wynn R, Greil J, Sykora KW, Matthes-Martin S, Fuhrer M, O'Meara A, Toporski J, Sedlacek P, Schlegel PG, Ehlert K, Fasth A, Winiarski J, Arvidson J, Mauz-Korholz C, Ozsahin H, Schrauder A, Bader P, Massaro J, D'Agostino R, Hoyle M, Iacobelli M, Debatin KM, Peters C, Dini G. Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation: an open-label, phase 3, randomised controlled trial. Lancet. 2012;379:1301-9. [PubMed: 22364685]
- Corbacioglu S, Honig M, Lahr G, Stohr S, Berry G, Friedrich W, Schulz AS. Stem cell transplantation in children with infantile osteopetrosis is associated with a high incidence of VOD, which could be prevented with defibrotide. Bone Marrow Transplant. 2006;38:547–53. [PubMed: 16953210]
- Delmonte OM, Baldin F, Ovchinsky N, Marquardsen F, Recher M, Notarangelo LD, Kosinski SM. Novel missense mutation in SP110 associated with combined immunodeficiency and advanced liver disease without VOD. J Clin Immunol. 2020;40:236–39. [PubMed: 31721003]
- Fraschilla I, Jeffrey KL. The speckled protein (SP) family: immunity's chromatin readers. Trends Immunol. 2020;41:572-85. [PMC free article: PMC8327362] [PubMed: 32386862]
- Ganaiem H, Eisenstein EM, Tenenbaum A, Somech R, Simanovsky N, Roscioli T, Weintraub M, Stepensky P. The role of hematopoietic stem cell transplantation in SP110 associated veno-occlusive disease with immunodeficiency syndrome. Pediatr Allergy Immunol. 2013;24:250–6. [PubMed: 23448538]
- Hsu K-S, Kao H-Y. PML: regulation and multifaceted function beyond tumor suppression. Cell Biosci. 2018;8:5:1-21. [PMC free article: PMC5753438] [PubMed: 29308184]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389-97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519-22. [PubMed: 28959963]
- Kishi M, Maeyama S, Ogata S, Koike J, Uchikoshi T. Hepatic veno-occlusive lesions in severe alcoholic hepatitis and alcoholic liver cirrhosis: a comparative histopathological study in autopsy cases. Alcohol Clin Exp Res. 1999;23:47S–51S. [PubMed: 10235278]
- Marquardsen FA, Baldin F, Wunderer F, Al-Herz W, Mikhael R, Lefranc G, Baz Z, Rezaee F, Hanna R, Kfir-Erenfeld S, Stepensky P, Meyer B, Jauch A, Bigler MB, Burgener AV, Higgins R, Navarini AA, Church JA, Chou J, Geha R, Notarangelo LD, Hess C, Berger CT, Bloch DB, Recher M. Detection of Sp110 by flow cytometry and application to screening patients for veno-occlusive disease with immunodeficiency. J Clin Immunol. 2017;37:707-14. [PMC free article: PMC6069968] [PubMed: 28825155]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Richardson PG, Smith AR, Triplett BM, Kernan NA, Grupp SA, Antin JH, Lehmann L, Miloslavsky M, Hume R, Hannah AL, Nejadnik B, Soiffer RJ. Earlier defibrotide initiation post-diagnosis of veno-occlusive disease/sinusoidal obstruction syndrome improves Day +100 survival following haematopoietic stem cell transplantation. Br J Haematol. 2017;178:112-8. [PMC free article: PMC5635854] [PubMed: 28444784]
- Roscioli T, Cliffe ST, Bloch DB, Bell CG, Mullan G, Taylor PJ, Sarris M, Wang J, Donald JA, Kirk EP, Ziegler JB, Salzer U, McDonald GB, Wong M, Lindeman R, Buckley MF. Mutations in the gene encoding the PML nuclear body protein Sp110 are associated with immunodeficiency and hepatic veno-occlusive disease. Nat Genet. 2006;38:620–2. [PubMed: 16648851]
- Ruga EM, Guariso G, Antiga LD, Guido M, Fassan M, Elia RD, Ziegler JB, Roscioli T, Cliffe ST, Buckley MF, Zancan L. Hepatic veno-occlusive disease with immunodeficiency syndrome: case report. 12th Meeting of the European Society for Immunodeficiencies, Budapest, Hungary. 2006.
- Srisirirojanakorn N, Finegold MJ, Gopalakrishna GS, Klish WJ. Hepatic veno-occlusive disease in ataxia-telangiectasia. J Pediatr. 1999;134:786–8. [PubMed: 10356154]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197-207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Wang T, Ong P, Roscioli T, Cliffe ST, Church JA. Hepatic veno-occlusive disease with immunodeficiency (VODI): first reported case in the U.S. and identification of a unique mutation in Sp110. Clin Immunol. 2012;145:102–7. [PubMed: 22982295]
- Watashi K, Hijikata M, Tagawa A, Doi T, Marusawa H, Shimotohno K. Modulation of retinoid signaling by a cytoplasmic viral protein via sequestration of Sp110b, a potent transcriptional corepressor of retinoic acid receptor, from the nucleus. Mol Cell Biol. 2003;23:7498–509. [PMC free article: PMC207568] [PubMed: 14559998]
- Wozniak LJ, Yang H-M, Lassman CR, Federman N, Wu SS. Extramedullary acute myelocytic leukemia following liver transplantation for VOD with immunodeficiency. J Pediatr Gastroenterol Nutr. 2011;53:346-9. [PubMed: 21865981]
Publication Details
Author Information and Affiliations
The Children's Hospital at Westmead
Sydney, Australia
Publication History
Initial Posting: February 21, 2007; Last Update: January 23, 2025.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2025 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Wong M. Hepatic Veno-Occlusive Disease with Immunodeficiency. 2007 Feb 21 [Updated 2025 Jan 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025.