Summary
Clinical characteristics.
Shprintzen-Goldberg syndrome (SGS) is characterized by: delayed motor and cognitive milestones and mild-to-moderate intellectual disability; craniosynostosis of the coronal, sagittal, or lambdoid sutures; distinctive craniofacial features; and musculoskeletal findings including olichostenomelia, arachnodactyly, camptodactyly, pectus excavatum or carinatum, scoliosis, joint hypermobility or contractures, pes planus, foot malposition, and C1-C2 spine malformation. Cardiovascular anomalies may include mitral valve prolapse, secundum atrial septal defect, and aortic root dilatation. Minimal subcutaneous fat, abdominal wall defects, and myopia are also characteristic findings.
Diagnosis/testing.
The diagnosis of SGS is established in a proband with a heterozygous pathogenic variant in SKI identified by molecular genetic testing.
Management.
Treatment of manifestations: Early intervention for developmental delay with placement in special education programs; standard management of cleft palate and craniosynostosis; surgical fixation may be necessary for cervical spine instability; routine management for scoliosis; surgical correction for pectus excavatum is rarely indicated; physiotherapy for joint contractures; clubfoot deformity may require surgical correction. If aortic dilatation is present, treatment with beta-adrenergic blockers or other medications should be considered in order to reduce hemodynamic stress; surgical intervention for aneurysms may be indicated; treatment of myopia as per ophthalmologist; surgical repair of abdominal hernias as indicated.
Prevention of secondary complications: Subacute bacterial endocarditis prophylaxis is recommended for dental work or other procedures for individuals with cardiac complications.
Surveillance: Developmental assessment with each visit; cervical spine evaluation and clinical evaluation for scoliosis as recommended by orthopedist; imaging per cardiologist familiar with this condition; ophthalmology exams as recommended by ophthalmologist.
Agents/circumstances to avoid: Contact sports; use of agents that stimulate the cardiovascular system; activities that may lead to joint pain and/or injury.
Genetic counseling.
SGS, caused by a heterozygous pathogenic variant in SKI, is an autosomal dominant disorder. Most individuals with SGS have unaffected parents, suggesting that the causative variant has occurred either as a de novo event in the affected individual or as a result of germline mosaicism in one of the parents. Affected sibs born to unaffected parents support the occurrence of germline mosaicism in some families with SGS. Once a SKI pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Diagnosis
Formal diagnostic criteria for Shprintzen-Goldberg syndrome (SGS) have not been established.
Suggestive Findings
SGS should be suspected in individuals with a combination of the following clinical (see Figure 1) and radiographic features:
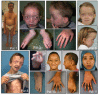
- Neurodevelopment. Hypotonia, delayed motor and cognitive milestones, mild-to-moderate intellectual disability
- Craniosynostosis usually involving the coronal, sagittal, or lambdoid sutures
- Craniofacial findings
- Dolichocephaly with or without scaphocephaly
- Tall or prominent forehead
- Hypertelorism
- Downslanting palpebral fissures
- Ocular proptosis
- Malar flattening
- High narrow palate with prominent palatine ridges
- Micrognathia and/or retrognathia
- Apparently low-set and posteriorly rotated ears
- Musculoskeletal findings
- Dolichostenomelia
- Arachnodactyly
- Camptodactyly
- Pectus excavatum or carinatum
- Scoliosis
- Joint hypermobility or contractures
- Pes planus
- Foot malposition/talipes equinovarus/club foot
- C1-C2 spine malformation
- Cardiovascular anomalies. Mitral valve prolapse/valvular anomalies, secundum atrial septal defect, aortic root dilatation
- Brain anomalies. Chiari I malformation
- Other. Minimal subcutaneous fat, abdominal wall defects, and myopia
Establishing the Diagnosis
The diagnosis of SGS is established in a proband with a heterozygous pathogenic (or likely pathogenic) variant in SKI identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of a heterozygous SKI variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of SGS is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other inherited disorders with craniosynostosis and additional congenital anomalies are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of SGS, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of SKI to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants.
- A multigene panel that includes SKI and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by craniosynostosis, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
If exome sequencing is not diagnostic – and particularly when evidence supports autosomal dominant inheritance – exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Shprintzen-Goldberg Syndrome
Clinical Characteristics
Clinical Description
To date, 44 individuals have been identified with a pathogenic variant in SKI [Carmignac et al 2012, Doyle et al 2012, Au et al 2014, Schepers et al 2015, Saito et al 2017, O'Dougherty et al 2019, Zhang et al 2019]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Select Features of Shprintzen-Goldberg Syndrome
Neurodevelopment. Motor and cognitive milestones are delayed and intellectual disability is usually mild to moderate. To date, three individuals with Shprintzen-Goldberg syndrome (SGS) and a confirmed SKI pathogenic variant were reported to have normal intelligence [Doyle et al 2012, Schepers et al 2015, Zhang et al 2019].
Craniosynostosis usually involves the coronal, sagittal, or lambdoid sutures. The sutures are fused at birth and craniosynostosis can usually be suspected from the abnormal skull shape. A single suture or multiple sutures may be involved [Au et al 2014]. Information on individuals with SGS who underwent surgery for craniosynostosis in unavailable.
Characteristic craniofacial features include hypertelorism, downslanting palpebral fissures, ocular proptosis, high narrow palate, micrognathia, and low-set posteriorly rotated ears. Cleft palate was reported in three of 17 individuals with SGS [Doyle et al 2012, Schepers et al 2015, O'Dougherty et al 2019]. Broad/bifid uvula has been reported in two of 12 individuals [Doyle et al 2012, O'Dougherty et al 2019].
Musculoskeletal. Arachnodactyly, camptodactyly, pectus deformities, and clubfeet are common features and are present at birth. Joint contractures are often present at birth or in the neonatal period, with the ankle joint most frequently affected [Au et al 2014, Saito et al 2017]. Joint hypermobility also occurs in individuals with SGS and is typically present from birth. Joint dislocation and instability reported by O'Dougherty et al [2019] included involvement of the left patella, thumb, and foot and C1-C2 instability. Au et al [2014] reported subluxing patellae. Pes planus becomes evident later in childhood; one or both feet may be affected. Scoliosis may be severe [Au et al 2014].
Cardiovascular. Aortic root dilatation was present in three of 18 affected individuals reported by Carmignac et al [2012]. In the report of Doyle et al [2012], however, eight of ten individuals with SGS and confirmed pathogenic variants in SKI had aortic root dilatation with or without mitral valve prolapse/incompetence. Surgery at age 16 years for aortic dilatation (aortic root dilatation with z score = 7.014) was reported in one individual with molecularly confirmed SGS [Carmignac et al 2012]. This individual also had vertebrobasilar and internal carotid tortuosity and a dilated pulmonary artery root. Among the affected individuals with molecularly confirmed SGS reported by Doyle et al [2012] one had arterial tortuosity and two had splenic artery aneurysm – one with spontaneous rupture.
Ocular. Myopia was reported in ten of 19 individuals [Carmignac et al 2012, Au et al 2014, O'Dougherty et al 2019]. Ectopia lentis has not been reported in the 16 individuals with SGS who were evaluated for this finding [Doyle et al 2012, Schepers et al 2015].
Minimal subcutaneous fat and/or marfanoid habitus was reported in nine of 20 individuals [Carmignac et al 2012, Au et al 2014].
Abdominal wall defects were reported in 14 of 23 individuals [Carmignac et al 2012, Au et al 2014, Schepers et al 2015, Saito et al 2017, O'Dougherty et al 2019].
Other
- Chiari I malformation (2/3 individuals) [Au et al 2014, O'Dougherty et al 2019]
- Cryptorchidism (1/1 male) [Saito et al 2017]
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been identified.
Penetrance
Penetrance is unknown.
Nomenclature
Goldberg-Shprintzen syndrome and Shprintzen-Goldberg omphalocele syndrome are separate syndromes, not related to SGS.
Other names that have been used to refer to SGS:
- Craniosynostosis with arachnodactyly and abdominal hernias
- Marfanoid-craniosynostosis syndrome
- Shprintzen-Goldberg craniosynostosis syndrome
- Shprintzen-Goldberg marfanoid syndrome
The term Furlong syndrome has been used to describe one individual with craniosynostosis, features of SGS, normal intelligence, and aortic enlargement. Adès et al [2006] reported on two individuals with a phenotype similar to Furlong syndrome. They had the same pathogenic missense variant in TGFBR1, making a diagnosis of Loeys-Dietz syndrome type 1 most likely [B Loeys, personal communication]. In the absence of analysis for pathogenic variants in the original individual described as having Furlong syndrome, the existence of this as a separate entity remains unclear.
Prevalence
SGS is a rare disorder and the prevalence is unknown. A SKI pathogenic variant has been identified in 44 individuals with SGS to date.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in SKI.
1p36 deletion syndrome (OMIM 607872). SKI is located in the critical region for the 1p36 deletion syndrome. It is currently unknown if SKI haploinsufficiency contributes to the phenotypic features associated with proximal 1p36 deletions. One individual with a small 1p36 deletion that included SKI, PRKCZ, and PEX10 had a phenotype that appeared to be distinct from SGS [Zhu et al 2013].
Differential Diagnosis
Loeys-Dietz syndrome (LDS) and Marfan syndrome (MFS). The phenotype of Shprintzen-Goldberg syndrome (SGS) is distinctive but shows some overlap with LDS and MFS (see Table 3). Distinguishing features of SGS include the following:
- Hypotonia and intellectual disability are rare findings in individuals with LDS and MFS but appear to be almost always present in those with SGS.
- Some of the distinctive radiographic findings in SGS are rarely found in individuals with either LDS or MFS. These include:
- C1/C2 abnormality (in 7/10 individuals with SGS) [Doyle et al 2012, Au et al 2014, Saito et al 2017, O'Dougherty et al 2019];
- Thirteen pairs of ribs (1/1) [O'Dougherty et al 2019];
- Chiari I malformation (2/3 ) [Au et al 2014, O'Dougherty et al 2019].
- Aortic root dilatation is less frequent in SGS than in LDS or MFS, but when present in individuals with SGS, it can be severe [Carmignac et al 2012]. One of the hallmarks of LDS is the occurrence of arterial tortuosity and aneurysms in arteries other than the aorta. Arterial tortuosity was found in two individuals with SGS; a further two individuals with SGS had splenic artery aneurysm [Carmignac et al 2012, Doyle et al 2012].
Table 3.
Comparison of Clinical Features of SGS, TGFBR1-/TGFBR2-LDS, and MFS
Other disorders
Table 4.
Disorders of Interest in the Differential Diagnosis of Shprintzen-Goldberg Syndrome
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Shprintzen-Goldberg syndrome (SGS), the evaluations summarized in Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 5.
Recommended Evaluations Following Initial Diagnosis in Individuals with Shprintzen-Goldberg Syndrome
Treatment of Manifestations
Management of SGS is best conducted through the coordinated input of a multidisciplinary team of specialists including a clinical geneticist, cardiologist, ophthalmologist, orthopedist, cardiothoracic surgeon, and craniofacial team.
Table 6.
Treatment of Manifestations in Individuals with Shprintzen-Goldberg Syndrome
Prevention of Secondary Complications
For individuals with cardiac complications, subacute bacterial endocarditis prophylaxis is recommended for dental work or other procedures expected to contaminate the bloodstream with bacteria.
Surveillance
Table 7.
Recommended Surveillance for Individuals with Shprintzen-Goldberg Syndrome
Agents/Circumstances to Avoid
The following should be avoided:
- Contact sports, which may lead to catastrophic complications in those with cardiovascular issues or cervical spine anomalies/instability
- Agents that stimulate the cardiovascular system, including routine use of decongestants
- Activities that cause joint pain or injury
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Shprintzen-Goldberg syndrome (SGS), caused by a heterozygous pathogenic variant in SKI, is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- To date, most probands diagnosed with SGS represent simplex cases (i.e., a single occurrence in a family) and are assumed to have the disorder as a result of a de novo pathogenic variant.
- Rarely, an individual diagnosed with SGS has the disorder as the result of an inherited pathogenic variant. Recurrence of SGS was reported in a three-generation family with SGS [Carmignac et al 2012] (family 3).
- Molecular genetic testing is recommended for the parents of a proband with an apparent de novo pathogenic variant.
- If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, possible explanations include a de novo pathogenic variant in the proband or germline mosaicism in a parent. Parental germline mosaicism could explain familial recurrence with unaffected parents. See Carmignac et al [2012] (family 4: individuals 8, 9, 10), Schepers et al [2015] (individuals 10, 11).
- The family history of some individuals diagnosed with SGS may appear to be negative because of a milder phenotypic presentation in an affected family member. Therefore, an apparently negative family history cannot be confirmed until appropriate clinical evaluation and/or molecular genetic testing has been performed.
- If the parent is the individual in whom the pathogenic variant first occurred, the parent may have somatic mosaicism for the variant and may be mildly/minimally affected.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband is heterozygous for the pathogenic variant identified in the proband, the risk to the sibs is 50%. Multigenerational familial recurrence of SGS is rare but has been reported [Carmignac et al 2012].
- If the proband has a known SKI pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental germline mosaicism [Carmignac et al 2012, Schepers et al 2015].
- If the parents have not been tested for the SKI pathogenic variant but are clinically unaffected, sibs are still presumed to be at increased risk for SGS because of the possibility of reduced penetrance in a heterozygous parent or parental germline mosaicism.
Offspring of a proband. Each child of an individual with SGS has a 50% chance of inheriting the pathogenic variant and having SGS.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the pathogenic variant, members of the parent's family may be at risk.
Related Genetic Counseling Issues
Considerations in families with an apparent de novo pathogenic variant. When neither parent of a proband with an autosomal dominant condition has the pathogenic variant identified in the proband or clinical evidence of the disorder, the pathogenic variant is likely de novo. However, non-medical explanations including alternate paternity or maternity (e.g., with assisted reproduction) and undisclosed adoption could also be explored.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the SKI pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Children's Craniofacial AssociationPhone: 800-535-3643Email: contactCCA@ccakids.com
- FACES: National Craniofacial AssociationPhone: 800-332-2373; 423-266-1632Email: info@faces-cranio.org
- National Institute of Neurological Disorders and Stroke (NINDS)Phone: 800-352-9424
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Shprintzen-Goldberg Syndrome: Genes and Databases
Table B.
OMIM Entries for Shprintzen-Goldberg Syndrome (View All in OMIM)
Molecular Pathogenesis
SKI encodes the nuclear protooncogene protein homolog of avian sarcoma viral (v-ski) oncogene, which functions as a repressor of TGF-β signaling. The SKI family of proteins negatively regulate SMAD-dependent TGF-β signaling by preventing nuclear translocation of the receptor-activated SMAD (R-SMAD)-SMAD4 complex and inhibiting TGF-β target gene output by competing with p300/CBP for SMAD binding and recruiting transcriptional repressor proteins, such as mSin3A and HDAC1.
The SKI oncogene is present in all cells, and is commonly active during development. It has a 728 amino-acid sequence with multiple domains and is expressed both inside and outside the cell. The different domains have different functions, including interaction with SMAD proteins.
Mechanism of disease causation. Cultured dermal fibroblasts from individuals with molecularly confirmed SGS showed enhanced activation of TGF-β signaling cascades and higher expression of TGF-β-responsive genes relative to control cells. Doyle et al [2012] concluded that increased TBF-β signaling is the mechanism underlying SGS.
TGF-β signaling is crucial to normal development and maintenance of various organs, including the vasculature. SKI exerts a negative regulatory effect on TGF-β signaling by interacting with other cellular partners, such as SMAD proteins and transcriptional co-regulators. All pathogenic variants of SKI are located in exon 1 and cluster into two domains: the R-SMAD binding domain and daschund homology domain (DHD). Zebrafish studies indicate that SKI plays crucial roles in aortic morphogenesis and homeostasis, and that aortic aneurysm formation in individujals with SGS is a result of increased TGF-β signaling, similar to aortopathies in Marfan syndrome and Loeys-Dietz syndrome [Takeda et al 2018]. For a review of the regulatory effects of transcriptional cofactors Ski (Sloan-Kettering Institute) and SnoN (Ski novel) on the TGF-β signaling pathway in health and disease, see Tecalco-Cruz et al [2018].
SKI-specific laboratory technical considerations. The majority of pathogenic variants are missense and are located in exon 1 of SKI [Carmignac et al 2012]. A mutational hot spot has been identified in the R-SMAD domain of exon 1 with 24 (73%) of 33 unrelated individuals having a pathogenic variant within a stretch of five residues (from Ser31 to Pro35). A smaller number of individuals with pathogenic variants in the DHD domain have been reported, suggesting a second mutational hotspot in this domain [Zhang et al 2019].
Chapter Notes
Revision History
- 9 April 2020 (sw) Comprehensive update posted live
- 13 June 2013 (me) Comprehensive update posted live
- 16 November 2010 (me) Comprehensive update posted live
- 13 January 2006 (me) Review posted live
- 2 December 2004 (mtg) Original submission
References
Literature Cited
- Adès LC, Sullivan K, Biggin A, Haan EA, Brett M, Holman KJ, Dixon J, Robertson S, Holmes AD, Rogers J, Bennetts B. FBN1, TGFBR1, and the Marfan-craniosynostosis/mental retardation disorders revisited. Am J Med Genet A. 2006;140:1047–58. [PubMed: 16596670]
- Au PY, Racher HE, Graham JM Jr, Kramer N, Lowry RB, Parboosingh JS, Innes AM, et al. De novo exon 1 missense mutations of SKI and Shprintzen-Goldberg syndrome: two new cases and clinical review. Am J Med Genet A. 2014;164A:676–84. [PubMed: 24357594]
- Carmignac V, Thevenon J, Adès L, Callewaert B, Julia S, Thauvin-Robinet C, Gueneau L, Courcet JB, Lopez E, Holman K, Renard M, Plauchu H, Plessis G, De Backer J, Child A, Arno G, Duplomb L, Callier P, Aral B, Vabres P, Gigot N, Arbustini E, Grasso M, Robinson PN, Goizet C, Baumann C, Di Rocco M, Sanchez Del Pozo J, Huet F, Jondeau G, Collod-Beroud G, Beroud C, Amiel J, Cormier-Daire V, Rivière JB, Boileau C, De Paepe A, Faivre L. In-frame mutations in exon 1 of SKI cause dominant Shprintzen-Goldberg syndrome. Am J Hum Genet. 2012;91:950–7. [PMC free article: PMC3487125] [PubMed: 23103230]
- Doyle AJ, Doyle JJ, Bessling SL, Maragh S, Lindsay ME, Schepers D, Gillis E, Mortier G, Homfray T, Sauls K, Norris RA, Huso ND, Leahy D, Mohr DW, Caulfield MJ, Scott AF, Destrée A, Hennekam RC, Arn PH, Curry CJ, Van Laer L, McCallion AS, Loeys BL, Dietz HC. Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat Genet. 2012;44:1249–54. [PMC free article: PMC3545695] [PubMed: 23023332]
- O'Dougherty GR, Fulkerson DH, Kern M, Haldar K, Calhoun B. Complications of insufficient dura and blood loss during surgical intervention in Shprintzen-Goldberg syndrome: a case report. Am J Case Rep. 2019;20:1159–69. [PMC free article: PMC6698069] [PubMed: 31391415]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Saito T, Nakane T, Yagasaki H, Naito A, Sugita K. Shprintzen-Goldberg syndrome associated with first cervical vertebra defects. Pediatr Int. 2017;59:1098–100. [PubMed: 28857439]
- Schepers D, Doyle AJ, Oswald G, Sparks E, Myers L, Willems PJ, Mansour S, Simpson MA, Frysira H, Maat-Kievit A, Van Minkelen R, Hoogeboom JM, Mortier GR, Titheradge H, Brueton L, Starr L, Stark Z, Ockeloen C, Lourenco CM, Blair E, Hobson E, Hurst J, Maystadt I, Destrée A, Girisha KM, Miller M, Dietz HC, Loeys B, Van Laer L. The SMAD-binding domain of SKI: a hotspot for de novo mutations causing Shprintzen-Goldberg syndrome. Eur J Hum Genet. 2015;23:224–8. [PMC free article: PMC4297897] [PubMed: 24736733]
- Takeda N, Hara H, Fujiwara T, Kanaya T, Maemura S, Komuro I. TGF-β signaling-related genes and thoracic aortic aneurysms and dissections. Int J Mol Sci. 2018;19:2125. [PMC free article: PMC6073540] [PubMed: 30037098]
- Tecalco-Cruz AC, Ríos-López DG, Vázquez-Victorio G, Rosales-Alvarez RE, Macías-Silva M. Transcriptional cofactors Ski and SnoN are major regulators of the TGF-β/Smad signaling pathway in health and disease. Signal Transduct Target Ther. 2018;3:15. [PMC free article: PMC5992185] [PubMed: 29892481]
- Yauy K, Tran Mau-Them F, Willems M, Coubes C, Blanchet P, Herlin C, Taleb Arrada I, Sanchez E, Faure JM, Le Gac MP, Prodhomme O, Boland A, Meyer V, Rivière JB, Duffourd Y, Deleuze JF, Guignard T, Captier G, Barat-Houari M, Genevieve D. B3GAT3-related disorder with craniosynostosis and bone fragility due to a unique mutation. Genet Med. 2018;20:269–74. [PubMed: 28771243]
- Zhang L, Xu X, Sun K, Sun J, Wang Y, Liu Y, Yang N, Tao C, Cai B, Shi G, Zhang F, Shi J. A de novo mutation in DHD domain of SKI causing spina bifida with no craniofacial malformation or intellectual disability. Am J Med Genet A. 2019;179:936–9. [PubMed: 30883014]
- Zhu X, Zhang Y, Wang J, Yang JF, Yang YF, Tan ZP. 576 kb deletion in 1p36.33-p36.32 containing SKI is associated with limb malformation, congenital heart disease and epilepsy. Gene. 2013;528:352–5. [PubMed: 23892090]
Publication Details
Author Information and Affiliations
Our Lady's Children's Hospital, Crumlin
Dublin, Ireland
Publication History
Initial Posting: January 13, 2006; Last Update: April 9, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Greally MT. Shprintzen-Goldberg Syndrome. 2006 Jan 13 [Updated 2020 Apr 9]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
