Summary
Clinical characteristics.
Laing distal myopathy is characterized by early-onset weakness (usually before age 5 years) that initially involves the dorsiflexors of the ankles and great toes and then the finger extensors, especially those of the third and fourth fingers. Weakness of the neck flexors is seen in most affected individuals and mild facial weakness is often present. After distal weakness has been present for more than ten years, mild proximal weakness may be observed. Life expectancy is normal.
Diagnosis/testing.
The diagnosis of Laing distal myopathy is established in a proband with suggestive findings and a heterozygous pathogenic variant in MYH7 identified by molecular genetic testing.
Management.
Treatment of manifestations: Physiotherapy to prevent or treat tightening of the Achilles tendon is helpful. In more advanced cases, lightweight splinting of the ankle (e.g., with an ankle-foot orthosis) can be useful. Standard medical treatment under the supervision of a cardiologist is recommended for cardiomyopathy; surgical stabilization of the spine is used to treat kyphoscoliosis.
Surveillance: Annual neurologic examination; repeat electrocardiogram and echocardiogram if symptoms of cardiac insufficiency occur; regular evaluation for scoliosis/kyphoscoliosis (especially during rapid growth); respiratory assessment if symptoms suggest sleep-related respiratory insufficiency and obstructive sleep apnea.
Genetic counseling.
Laing distal myopathy is an autosomal dominant disorder. Approximately 65%-70% of affected individuals have an affected parent; de novo pathogenic variants in MYH7 account for 30%-35% of individuals with Laing distal myopathy. If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%. Once the MYH7 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for Laing distal myopathy are possible.
Diagnosis
No consensus clinical diagnostic criteria for Laing distal myopathy have been published.
Suggestive Findings
Laing distal myopathy should be considered in individuals with the following findings [Hedera et al 2003, Lamont et al 2006, Lamont et al 2014].
Clinical findings
- Initial weakness of the great toe and ankle dorsiflexors, eventually leading to a high-stepping gait and secondary tightening of the Achilles tendon. Onset is usually before age five years, but may be later (into the 6th decade).
- Subsequent weakness of the finger extensors (onset from months to 3 decades after lower-limb weakness), with sparing of the thumb, and often accompanied by an action tremor of the hands
- Mild involvement of the facial musculature, particularly of the orbicularis oculi and oris muscles
- Early weakness of neck flexion in most families
- Very slow progression of weakness with gradual involvement of the proximal leg and trunk muscles. With early onset, a wheelchair may eventually be required for mobility.
Laboratory findings
- Serum creatine kinase concentration is usually normal, but may in rare cases be as high as eight times the upper limit of normal.
- Nerve conduction studies are normal.
- Electromyographic findings are nonspecific, with occasional fibrillation potentials but no prolonged or large motor unit potentials [Zimprich et al 2000].
Family history is consistent with autosomal dominant inheritance (e.g., affected males and females in multiple generations). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of Laing distal myopathy is established in a proband with suggestive findings and a heterozygous pathogenic variant in MYH7 identified by molecular genetic testing (see Table 1).
Note: Identification of a heterozygous MYH7 variant of uncertain significance does not establish or rule out the diagnosis of this disorder.
Because the phenotype of Laing distal myopathy can be indistinguishable from many other inherited disorders with muscle weakness, recommended molecular genetic testing approaches include use of a multigene panel or comprehensive genomic testing.
Note: Single-gene testing (sequence analysis of MYH7, followed by gene-targeted deletion/duplication analysis) is rarely useful and typically NOT recommended.
- A multigene panel for distal myopathy that includes MYH7 and other genes of interest (see Differential Diagnosis) as described in Beecroft et al [2020] is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
- Comprehensive genomic testing, which does not require the clinician to determine which gene(s) are likely involved, is another good option. Exome sequencing is most commonly used; genome sequencing is also possible.
Table 1.
Molecular Genetic Testing Used in Laing Distal Myopathy
Clinical Characteristics
Clinical Description
Laing distal myopathy is characterized by muscle weakness and atrophy beginning in the lower legs [Lamont et al 2006]. Onset is often before age five years. In a few children, onset has been so early as to delay walking. In two families, weakness was not recognized until the teenage years [Zimprich et al 2000, Hedera et al 2003, Lamont et al 2006]. In one family with 20 affected members, onset of lower-limb weakness occurred between early childhood and the fourth decade [Lamont et al 2014]. Onset as late as the sixth decade has been described [Hara et al 2019].
More than 200 individuals have been identified with a pathogenic variant in MYH7 associated with Laing distal myopathy. The following description of the phenotypic features associated with this condition is based on the reports of Lamont et al [2014], Fiorillo et al [2016], and Dabaj et al [2018].
Table 2.
Laing Distal Myopathy: Frequency of Select Features
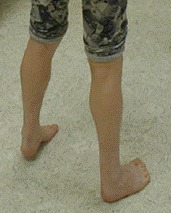
Lower leg weakness follows a typical sequence: initially dorsiflexion of the ankle and great toe is affected and leads to a high-stepping gait, dropped big toe, and secondary tightening of the Achilles tendon (see Figure 1).
Figure 1.
Early development of anterior compartment weakness has led to marked tightening of the Achilles tendon bilaterally, with the affected individual unable to place his heels on the ground.
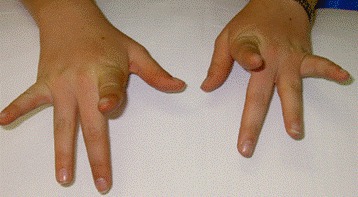
Weakness of finger extensors develops between months and several decades after the onset of leg weakness [Lamont et al 2014]. The third and fourth fingers appear to be more severely affected than the other fingers (see Figure 2), although any of the fingers can be affected. The thumb is spared. Weakness of the finger extensors is often accompanied by a postural and action tremor of the hands.
Figure 2.
Individual with Laing distal myopathy attempting to extend her second to fifth fingers. Note marked weakness of third- and fourth-finger extension.
Mild facial weakness is often present, leading to inability to bury the eyelashes completely when closing the eyes tightly, and inability to keep the lips pursed against resistance. One affected individual has a mild Bell's phenomenon.
Weakness of neck flexion, seen in all affected individuals, is usually early in onset, though weakness of neck flexion did not manifest in one family until the sixth decade. In most affected individuals and sites, the weakness is symmetric.
Proximal weakness. After distal weakness has been present for more than ten years, mild proximal weakness occurs, with a slight Trendelenburg gait and mild scapular winging (see Figure 3). Axial musculature may be mildly weak as well (manifesting as, e.g., inability to do a sit-up).

Figure 3.
Mild scapular winging and weakness develops later.
Progression is usually extremely slow; however, in one person the weakness became generalized and a wheelchair was required for mobility by age 15 years [Lamont et al 2014].
Spinal manifestations, which can include kyphoscoliosis, spinal rigidity, and spinal extensor muscle contractures, occur in one third of individuals and can vary within a family [Feinstein-Linial et al 2016, Fiorillo et al 2016]. Severe axial involvement with scoliosis, cervical hyperextension, and bent spine has been described [Dabaj et al 2018].
Cardiac problems are common. In their review of 88 affected individuals from 22 families, Lamont et al [2014] reported cardiac involvement ranging from hypertrophic cardiomyopathy with onset from birth to the third decade of life, to dilated cardiomyopathy with onset from birth to the second decade of life. In an earlier report, a father and son in one family developed a dilated cardiomyopathy for which no other cause was found [Hedera et al 2003].
Respiratory issues, present in approximately 40% of individuals in the form of reduced forced vital capacity, are not usually life threatening [Lamont et al 2014]. Sleep apnea or sleep-related respiratory insufficiency may develop [Yu et al 2020].
CNS involvement with white matter lesions and epilepsy has been described in a single family including three of 14 family members over three generations [Lefter et al 2015].
Muscle pathology in Laing distal myopathy is highly variable [Lamont et al 2006, Lamont et al 2014].
- The most common myopathic feature is excessive variation in fiber size, with either type 1 or type 2 fibers involved.Fiber type predominance is common. In one large family, ten of 14 muscle biopsies showed abnormally small type 1 fibers with type 1 predominance, fulfilling criteria for congenital fiber-type disproportion [Muelas et al 2010].
- Another common finding is core pathology of either central cores or multiminicores, with or without subsarcolemmal hyaline bodies [Cullup et al 2012, Negrão et al 2020].
- Other reported findings:
- Excessive central nucleation and mild necrosis and regeneration
- Fatty replacement in "end-stage" muscles
- Cytoplasmic bodies and myofibrillar-like myopathy features [Tasca et al 2012]
- Spheroid cytoplasmic body-like inclusions with a moth-eaten appearance [Hara et al 2019]
- Inflammatory myopathy with rimmed vacuoles resembling inclusion body myositis [Roda et al 2014]
- Immunohistochemical staining for slow and fast myosin demonstrating co-expression of both isoforms in some muscle fibers, possibly indicating a switch from fiber type 1 to fiber type 2 [Lamont et al 2006]
Genotype-Phenotype Correlations
Laing distal myopathy may be caused by different types of variants in the distal myosin tail. These include missense changes that insert proline, or cause charge changes or deletion or insertion of amino acids [Lamont et al 2014]. Charge reversal pathogenic variants in MYH7 including p.Glu1801Lys, p.Glu1856Lys, and p.Glu1914Lys can be associated with a Laing distal myopathy phenotype combined with cardiomyopathy [Udd 2009, Finsterer et al 2014a, Finsterer et al 2014b, Lamont et al 2014]. It has also been shown that missense pathogenic variants to proline (p.Arg1608Pro) and amino acid deletions (p.Leu1793del, p.Lys1617del) or insertions can also be associated with a combined distal myopathy/cardiomyopathy phenotype [Lamont et al 2014].
Penetrance
Penetrance appears to be at least 85%.
Muelas et al [2010] reported a large Spanish family in which the age of onset ranged from birth to the sixth decade; 15% of family members with the pathogenic variant were reported to be asymptomatic. (Note, however, that individual ages at the time of reporting were not clearly stated.)
In one apparent instance of a de novo pathogenic variant, the supposedly unaffected father was found to have somatic mosaicism; however, when examined, he did have mild weakness [Lamont et al 2014].
Nomenclature
The following alternate terms for Laing distal myopathy are no longer in use or are too nonspecific to be useful:
- Early-onset chromosome 14-linked distal myopathy (Laing)
- Autosomal dominant distal muscular dystrophy
- Infantile autosomal dominant distal myopathy
- Autosomal dominant distal myopathy (a nonspecific term that could apply to other distal myopathies such as tibial muscular dystrophy)
- Gowers myopathy
Prevalence
The prevalence of Laing distal myopathy is unknown. It is thought to be the most common distal myopathy worldwide [B Udd, personal communication], accounting for approximately 50% of early-onset distal myopathy [Author, personal observation]. The frequency of de novo pathogenic variants would also suggest a relatively high prevalence.
Laing distal myopathy has been reported in most populations [Park et al 2013, Lamont et al 2014, Hara et al 2019] and does not appear to be more prevalent in any specific populations [Author, personal observation].
Genetically Related (Allelic) Disorders
Other phenotypes associated with germline pathogenic variants in MYH7 are summarized in Table 3.
Table 3.
MYH7 Allelic Disorders
Differential Diagnosis
Other disorders to consider in the differential diagnosis of Laing distal myopathy are indicated in this section.
Congenital Myopathy
The early onset of Laing distal myopathy means that any of the milder congenital myopathies may be a differential diagnosis (see Table 4a).
Table 4a.
Congenital Myopathies of Interest in the Differential Diagnosis of Laing Distal Myopathy
Distal Myopathies
The other major group in the differential diagnosis of Laing distal myopathy is distal myopathies (see Table 4b).
Table 4b.
Distal Myopathies of Interest in the Differential Diagnosis of Laing Distal Myopathy
Charcot-Marie-Tooth (CMT) hereditary neuropathy, a group of disorders characterized by a chronic motor and sensory polyneuropathy, also commonly features foot drop and thus may be considered in the differential diagnosis. Laing distal myopathy is frequently mistakenly diagnosed as CMT.
- Muscle weakness in CMT is often associated with mild-to-moderate distal sensory loss. Although usually described as "painless," the neuropathy can be painful. Sensory loss can most easily be demonstrated by a decreased appreciation of vibration, but can also include impaired sensation of pain/pinprick, temperature, and joint position. More than 80 genes are associated with CMT.
- One aid to differential diagnosis between Laing distal myopathy and CMT: unlike in CMT, in Laing distal myopathy the extensor digitorum brevis muscles are preserved [Lamont et al 2014].
Management
No clinical practice guidelines for Laing distal myopathy have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Laing distal myopathy, the evaluations summarized in Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 5.
Recommended Evaluations Following Initial Diagnosis in Individuals with Laing Distal Myopathy
Treatment of Manifestations
Table 6.
Treatment of Manifestations in Individuals with Laing Distal Myopathy
Surveillance
Table 7.
Recommended Surveillance for Individuals with Laing Distal Myopathy
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk relatives of an affected individual in order to identify as early as possible those who would benefit from physiotherapy and surveillance for cardiomyopathy. Evaluations can include:
- Molecular genetic testing if the pathogenic variant in the family is known;
- Evaluations for muscle weakness and secondary contractures if the pathogenic variant in the family is not known.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Laing distal myopathy is an autosomal dominant disorder.
Risk to Family Members
Parents of a proband
- Approximately 65%-70% of individuals diagnosed with Laing distal myopathy have an affected parent.
- De novo pathogenic variants in MYH7 account for 30%-35% of individuals with Laing distal myopathy.
- Recommendations for the evaluation of parents of a proband who appears to be the only affected family member (i.e., a simplex case) include full history, examination looking for weakness and secondary contractures, and molecular genetic testing for the MYH7 pathogenic variant identified in the proband.
- If the pathogenic variant identified in the proband is not identified in either parent, the following possibilities should be considered:
- The proband has a de novo pathogenic variant. Note: A pathogenic variant is reported as "de novo" if: (1) the pathogenic variant found in the proband is not detected in parental DNA; and (2) parental identity testing has confirmed biological maternity and paternity. If parental identity testing is not performed, the variant is reported as "assumed de novo" [Richards et al 2015].
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism.* Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism.* A parent with somatic and germline mosaicism for an MYH7 pathogenic variant may be mildly/minimally affected. One such family has been reported [Lamont et al 2014].
- The family history of some individuals diagnosed with Laing distal myopathy may appear to be negative because of failure to recognize the disorder in family members, early death of the parent before the onset of symptoms, or late onset of the disorder in the affected parent. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has demonstrated that neither parent is heterozygous for the pathogenic variant identified in the proband.
Sibs of a proband. The risk to the sibs of the proband depends on the clinical/genetic status of the proband's parents:
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%. Penetrance of Laing distal myopathy is approximately 85%; it is therefore 85% likely that a sib who inherits a familial pathogenic variant will have clinical manifestations of the disorder. Considerable intrafamilial variation in severity has been described in Laing distal myopathy; in one family, some heterozygous individuals were asymptomatic while others required a wheelchair for mobility [Muelas et al 2010].
- If the proband has a known MYH7 pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental mosaicism [Lamont et al 2014].
- If the parents have not been tested for the MYH7 pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for Laing distal myopathy because of the possibility of late onset of the disorder in a heterozygous parent or parental germline mosaicism.
Offspring of a proband. Each child of an individual with Laing distal myopathy has a 50% chance of inheriting the MYH7 pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected and/or is known to have the MYH7 pathogenic variant, his or her family members are at risk.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the MYH7 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for Laing distal myopathy are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Muscular Dystrophy Association (MDA) - USAPhone: 833-275-6321Email: ResourceCenter@mdausa.org
- Muscular Dystrophy CanadaCanadaPhone: 800-567-2873Email: info@muscle.ca
- Muscular Dystrophy UKUnited KingdomPhone: 0800 652 6352
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Laing Distal Myopathy: Genes and Databases
Table B.
OMIM Entries for Laing Distal Myopathy (View All in OMIM)
Molecular Pathogenesis
The MYH7 variants that cause Laing distal myopathy lie in exons of the gene that code for the myosin rod domain [Meredith et al 2004, Lamont et al 2014]. They include deletions or insertions of amino acid residues, missense variants to proline and missense changes that produce a charge reversal, such as glutamate to lysine [Lamont et al 2014].
The tail of a myosin molecule forms an alpha-helical coiled coil with the tail of another myosin molecule and through this process forms a dimer [McLachlan & Karn 1982]. In order for an amino acid chain to form a coiled coil, the amino acids must have a particular seven- (heptad – a,b,c,d,e,f,g) amino-acid repeating structure with only certain types of amino acid residues allowed in each of the positions – for example, hydrophobic residues at positions a and d – a structure investigated by Francis Crick before he worked out the structure of DNA [Crick 1953, McLachlan & Karn 1982, Tajsharghi & Oldfors 2013]. Any variants that alter the heptad repeat, such as deletion or insertion of an amino acid, will affect the ability of the amino-acid chain to form a coiled coil and tend to destabilize the formation of the myosin dimer.
Similarly, proline residues are not compatible with a coiled coil since they introduce a kink in protein structure [O'Neil & DeGrado 1990]. There are no prolines normally in the myosin tail [McLachlan & Karn 1982], with the last invariant proline at the junction between the myosin head and the myosin tail [Achal et al 2016].
Thus, most of the MYH7 variants associated with Laing distal myopathy would apparently interfere with the formation of the myosin tail coiled coil [Buvoli et al 2012].
The missense variants causing large charge changes are different. There is a pattern of charge changes along the length of the myosin rod, which plays a role in the formation of the thick filament [McLachlan & Karn 1982]. The missense variants in the myosin rod associated with Laing distal myopathy that cause a charge reversal such as p.Glu1856Lys or p.Glu1914Lys may thus interfere with the formation of the thick filament.
The pathogenic mechanism by which these effects on coiled coil or thick filament formation ultimately have the effects on specific muscles seen in Laing distal myopathy, such as early and severe involvement of the tibialis anterior, remains a complete mystery. As MYH7 protein is present in every slow muscle fiber in every muscle in the human body and in the heart, why then is the pattern of weakness restricted? It may not be possible to model the effect of Laing distal myopathy myosin variants in any animal other than human beings.
Mechanism of disease causation. Laing distal myopathy occurs via a dominant negative mechanism where the mutated myosin molecule affects interaction with the normal proteins of the thick filament.
MYH7-specific laboratory technical considerations. There are no technical issues with analysis of MYH7 using next-generation sequencing. The targeted gene panels described by Beecroft et al [2020] give 100% coverage of the coding exons of MYH7 [Mark Davis, PhD, personal communication].
Table 8.
Notable MYH7 Pathogenic Variants
Chapter Notes
Author Notes
Nigel Laing's work focuses on disease gene discovery, development and implementation of improved molecular diagnostics, preclinical investigation of potential treatments for selected genetic muscle diseases, and reproductive genetic carrier screening. He has a research professorship in the Centre for Medical Research, University of Western Australia, located in the Harry Perkins Institute of Medical Research, and a senior medical scientist position within the Neurogenetic Unit, Department of Diagnostic Genomics, PathWest Laboratory Medicine, Health Department of Western Australia.
Websites:
research-repository.uwa.edu.au
www.perkins.org.au
Phillipa Lamont's work focuses on clinical investigation and research of neuromuscular disorders, disease gene discovery, and clinical trials. Professor Lamont heads the Neurogenetic Clinic at Royal Perth Hospital in Perth, Western Australia, which provides a statewide service for genetic neurologic conditions.
Acknowledgments
NGL is supported by Australian National Health and Medical Research Council Fellowship APP1117510.
Author History
Nigel G Laing, PhD (2006-present)
Phillipa Lamont, MBBS, PhD (2006-present)
William Wallefeld, BSc (Hons); University of Western Australia (2010-2015)
Revision History
- 4 February 2021 (ha) Comprehensive update posted live
- 12 March 2015 (me) Comprehensive update posted live
- 17 June 2010 (me) Comprehensive update posted live
- 17 October 2006 (me) Review posted live
- 6 September 2006 (nl) Original submission
References
Literature Cited
- Achal M, Trujillo AS, Melkani GC, Farman GP, Ocorr K, Viswanathan MC, Kaushik G, Newhard CS, Glasheen BM, Melkani A, Suggs JA, Moore JR, Swank DM, Bodmer R, Cammarato A, Bernstein SI. A restrictive cardiomyopathy mutation in an invariant proline at the myosin head/rod junction enhances head flexibility and function, yielding muscle defects in drosophila. J Mol Biol. 2016;428:2446–61. [PMC free article: PMC4884507] [PubMed: 27107639]
- Beecroft SJ, van der Locht JM, Ottenheim CA, Sewry CA, Mohammed S, Ryan MM, Woodcock IR, Sanders L, Gooding R, Davis MR, Oates EC, Laing NG, Ravenscroft G, McLean CA, Jungbluth H. Recessive MYH7-related myopathy in 2 families. Neuromuscul Disord. 2019;29:456–67. [PubMed: 31130376]
- Beecroft SJ, Yau KS, Allcock RJN, Mina K, Gooding R, Faiz F, Atkinson VJ, Wise C, Sivadorai P, Trajanoski D, Kresoje N, Ong R, Duff RM, Cabrera-Serrano M, Nowak KJ, Pachter N, Ravenscroft G, Lamont PJ, Davis MR, Laing NG. Targeted gene panel use in 2249 neuromuscular patients: the Australasian referral center experience. Ann Clin Transl Neurol. 2020;7:353–62. [PMC free article: PMC7086001] [PubMed: 32153140]
- Buvoli M, Buvoli A, Leinwand LA. Effects of pathogenic proline mutations on myosin assembly. J Mol Biol. 2012;415:807–18. [PMC free article: PMC3267876] [PubMed: 22155079]
- Carbonell-Corvillo P, Tristán-Clavijo E, Cabrera-Serrano M, Servián-Morilla E, García-Martín G, Villarreal-Pérez L, Rivas-Infante E, Area-Gómez E, Chamorro-Muñoz MI, Gil-Gálvez A, Miranda-Vizuete A, Martinez-Mir A, Laing N, Paradas C. A novel MYH7 founder mutation causing Laing distal myopathy in Southern Spain. Neuromuscul Disord. 2018;28:828–36. [PubMed: 30166250]
- Crick FHC. The packing of a-helices: simple coiled coils. Acta Crystallographica. 1953;6:689–97.
- Cullup T, Lamont PJ, Cirak S, Damian MS, Wallefeld W, Gooding R, Tan SV, Sheehan J, Muntoni F, Abbs S, Sewry CA, Dubowitz V, Laing NG, Jungbluth H. Mutations in MYH7 cause multi-minicore disease with variable cardiac involvement. Neuromuscul Disord. 2012;22:1096–104. [PubMed: 22784669]
- Dabaj I, Carlier RY, Gómez-Andrés D, Neto OA, Bertini E, D'amico A, Fattori F. PéRéon Y, Castiglioni C, Rodillo E, Catteruccia M, Guimarães JB, Oliveira ASB, Reed UC, Mesrob L, Lechner D, Boland A, Deleuze JF, Malfatti E, Bonnemann C, Laporte J, Romero N, Felter A, Quijano-Roy S, Moreno CAM, Zanoteli E. Clinical and imaging hallmarks of the MYH7-related myopathy with severe axial involvement. Muscle Nerve. 2018;58:224–34. [PubMed: 29624713]
- Feinstein-Linial M, Buvoli M, Buvoli A, Sadeh M, Dabby R, Straussberg R, Shelef I, Dayan D, Leinwand LA, Birk OS. Two novel MYH7 proline substitutions cause Laing distal myopathy-like phenotypes with variable expressivity and neck extensor contracture. BMC Med Genet. 2016;17:57. [PMC free article: PMC4982306] [PubMed: 27519903]
- Finsterer J, Brandau O, Stollberger C, Wallefeld W, Laing NG, Laccone F. Distal myosin heavy chain-7 myopathy due to the novel transition c.5566G>A (p.E1856K) with high interfamilial cardiac variability and putative anticipation. Neuromuscul Disord. 2014a;24:721–5. [PubMed: 24953931]
- Finsterer J, Stollberger C, Brandau O, Laccone F, Bichler K, Laing NG. 2014b. Novel MYH7 mutation associated with mild myopathy but life-threatening ventricular arrhythmias and noncompaction. Int J Cardiol. 2014b;173:532–5. [PubMed: 24726209]
- Fiorillo C, Astrea G, Savarese M, Cassandrini D, Brisca G, Trucco F, Pedemonte M, Trovato R, Ruggiero L, Vercelli L, D'Amico A, Tasca G, Pane M, Fanin M, Bello L, Broda P, Musumeci O, Rodolico C, Messina S, Vita GL, Sframeli M, Gibertini S, Morandi L, Mora M, Maggi L, Petrucci A, Massa R, Grandis M, Toscano A, Pegoraro E, Mercuri E, Bertini E, Mongini T, Santoro L, Nigro V, Minetti C, Santorelli FM, Bruno C, et al. MYH7-related myopathies: clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J Rare Dis. 2016;11:91. [PMC free article: PMC4936326] [PubMed: 27387980]
- Hackman P, Sarparanta J, Lehtinen S, Vihola A, Evilä A, Jonson PH, Luque H, Kere J, Screen M, Chinnery PF, Åhlberg G, Edström L, Udd B. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Ann Neurol. 2013;73:500–9. [PubMed: 23401021]
- Hara K, Miyata H, Nishino I. Rinsho Shinkeigaku. 2019;59:823–8. [A case of Japanese Laing type distal myopathy with a mutation in MYH7 gene.] [PubMed: 31761835]
- Hedera P, Petty EM, Bui MR, Blaivas M, Fink JK. The second kindred with autosomal dominant distal myopathy linked to chromosome 14q: genetic and clinical analysis. Arch Neurol. 2003;60:1321–5. [PubMed: 12975303]
- Lamont PJ, Udd B, Mastaglia FL, de Visser M, Hedera P, Voit T, Bridges LR, Fabian V, Rozemuller A, Laing NG. Laing early onset distal myopathy: slow myosin defect with variable abnormalities on muscle biopsy. J Neurol Neurosurg Psychiatry. 2006;77:208–15. [PMC free article: PMC2077563] [PubMed: 16103042]
- Lamont PJ, Wallefeld W, Hilton-Jones D, Udd B, Argov Z, Barboi AC, Bonneman C, Boycott KM, Bushby K, Connolly AM, Davies N, Beggs AH, Cox GF, Dastgir J, DeChene ET, Gooding R, Jungbluth H, Muelas N, Palmio J, Penttilä S, Schmedding E, Suominen T, Straub V, Staples C, Van den Bergh PY, Vilchez JJ, Wagner KR, Wheeler PG, Wraige E, Laing NG. Novel mutations widen the phenotypic spectrum of slow skeletal/β-cardiac myosin (MYH7) distal myopathy. Hum Mutat. 2014;35:868–79. [PMC free article: PMC4112555] [PubMed: 24664454]
- Lefter S, Hardiman R, McLaughlin RL, Murphy SM, Farrell M, Ryan AM. A novel MYH7 Leu1453pro mutation resulting in Laing distal myopathy in an Irish family. Neuromuscul Disord. 2015;25:155–60. [PubMed: 25447691]
- McLachlan AD, Karn J. Periodic charge distributions in the myosin rod amino acid sequence match cross-bridge spacings in muscle. Nature. 1982;299:226–31. [PubMed: 7202124]
- Meredith C, Herrmann R, Parry C, Liyanage K, Dye DE, Durling HJ, Duff RM, Beckman K, de Visser M, van der Graaff MM, Hedera P, Fink JK, Petty EM, Lamont P, Fabian V, Bridges L, Voit T, Mastaglia FL, Laing NG. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause Laing early-onset distal myopathy (MPD1). Am J Hum Genet. 2004;75:703–8. [PMC free article: PMC1182058] [PubMed: 15322983]
- Muelas N, Hackman P, Luque H, Garcés-Sánchez M, Azorín I, Suominen T, Sevilla T, Mayordomo F, Gómez L, Martí P, María Millán J, Udd B, Vílchez JJ. MYH7 gene tail mutation causing myopathic profiles beyond Laing distal myopathy. Neurology. 2010;75:732–41. [PubMed: 20733148]
- Muelas N, Hackman P, Luque H, Suominen T, Espinos C, Garces-Sanchez M, Sevilla T, Azorin I, Millan J, Udd B, Vilchez J. Spanish MYH7 founder mutation of Italian ancestry causing a large cluster of Laing myopathy patients. Clin Genet. 2012;81:491–4. [PubMed: 21395566]
- Negrão L, Machado R, Lourenco M, Fernandez-Marmiesse A, Rebelo O. Laing distal myopathy with subsarcolemmal hyaline bodies caused by a novel variant in the MYH7 gene. Acta Myologica. 2020;39:24–8. [PMC free article: PMC7315894] [PubMed: 32607476]
- O'Neil KT, DeGrado WF. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science. 1990;250:646–51. [PubMed: 2237415]
- Pajusalu S, Talvik I, Noormets K, Talvik T, Poder H, Joost K, Puusepp S, Piirsoo A, Stenzel W, Goebel HH, Nikopensius T, Annilo T, Noukas M, Metspalu A, Ounap K, Reimand T. De novo exonic mutation in MYH7 gene leading to exon skipping in a patient with early onset muscular weakness and fiber-type disproportion. Neuromuscul Disord. 2016;26:236–9. [PubMed: 26782017]
- Park JM, Kim YJ, Yoo JH, Hong YB, Park JH, Koo H, Chung KW, Choi BO. A novel MYH7 mutation with prominent paraspinal and proximal muscle involvement. Neuromuscul Disord. 2013;23:580–6. [PubMed: 23707328]
- Pegoraro E, Gavassini BF, Borsato C, Melacini P, Vianello A, Stramare R, Cenacchi G, Angelini C. MYH7 gene mutation in myosin storage myopathy and scapulo-peroneal myopathy. Neuromuscul Disord. 2007;17:321–9. [PubMed: 17336526]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Roda RH, Schindler AB, Blackstone C, Mammen AL, Corse AM, Lloyd TE. Laing distal myopathy pathologically resembling inclusion body myositis. Ann Clin Transl Neurol. 2014;1:1053–8. [PMC free article: PMC4284131] [PubMed: 25574480]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Tajsharghi H, Oldfors A. Myosinopathies: pathology and mechanisms. Acta Neuropathologica. 2013;125:3–18. [PMC free article: PMC3535372] [PubMed: 22918376]
- Tasca G, Ricci E, Pentilla S, Monforte M, Giglio V, Ottavani P, Camstra G, Silvestri G, Udd B. New phenotype and pathology features in MYH7-related distal myopathy. Neuromuscul Disord. 2012;22:640–7. [PubMed: 22521714]
- Udd B. 165th ENMC International Workshop: distal myopathies 6-8th February 2009, Naarden, the Netherlands. Neuromuscul Disord. 2009;19:429–38. [PubMed: 19477645]
- von Tell D, Somer H, Udd B, Edstrom L, Borg K, Ahlberg G. Welander distal myopathy outside the Swedish population: phenotype and genotype. Neuromuscul Disord. 2002;12:544–7. [PubMed: 12117477]
- Yu M, Zhu Y, Lu Y, Lv H, Zhang W, Yuan Y, Wang Z. Clinical features and genotypes of Laing distal myopathy in a group of Chinese patients, with in-frame deletions of MYH7 as common mutations. Orphanet J Rare Dis. 2020;15:344. [PMC free article: PMC7727133] [PubMed: 33298082]
- Zimprich F, Djamshidian A, Hainfellner JA, Budka H, Zeitlhofer J. An autosomal dominant early adult-onset distal muscular dystrophy. Muscle Nerve. 2000;23:1876–9. [PubMed: 11102913]
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: October 17, 2006; Last Update: February 4, 2021.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Lamont P, Laing NG. Laing Distal Myopathy. 2006 Oct 17 [Updated 2021 Feb 4]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.