Summary
Clinical characteristics.
Retinoblastoma is a malignant tumor of the developing retina that occurs in children, usually before age five years. Retinoblastoma may be unifocal or multifocal. About 60% of affected individuals have unilateral retinoblastoma with a mean age of diagnosis of 24 months; about 40% have bilateral retinoblastoma with a mean age of diagnosis of 15 months. Heritable retinoblastoma is associated with susceptibility for retinoblastoma as well as non-ocular tumors.
Diagnosis/testing.
The diagnosis of retinoblastoma is usually established by examination of the fundus of the eye using indirect ophthalmoscopy. Imaging studies can be used to support the diagnosis and stage the tumor. Almost all retinoblastomas develop following biallelic inactivation of RB1 in a cone cell precursor in the developing retina. The diagnosis of heritable retinoblastoma is established in a proband with retinoblastoma or retinoma (benign potential precursor to retinoblastoma) and a family history of retinoblastoma or a heterozygous germline pathogenic variant in RB1 identified by molecular genetic testing. Epigenetic hypermethylation of the RB1 promotor also results in inactivation of RB1 but cannot be transmitted via the germline. Amplification of MYCN in a developing retinal cone cell can also result in retinoblastoma that is not thought to be heritable.
The following staging has been recommended for individuals with retinoblastoma and/or risk of heritable retinoblastoma to describe the genetic risk of a germline pathogenic variant in RB1:
- HX. Individual with unknown or insufficient evidence of a constitutional (germline) RB1 pathogenic variant
- H0. Individual who did not inherit a known familial germline RB1 pathogenic variant confirmed by molecular genetic testing
- (H0*. Individual with unilateral retinoblastoma or retinoma with no germline RB1 pathogenic variant identified on molecular genetic testing; residual risk of mosaicism is <1%.)
- H1. Individual with bilateral retinoblastoma, trilateral retinoblastoma (retinoblastoma with intracranial central nervous system midline embryonic tumor), retinoblastoma and a family history of retinoblastoma, or identification of a germline RB1 pathogenic variant
Management.
Treatment of manifestations: Early diagnosis and treatment of retinoblastoma can reduce morbidity and increase longevity; care is best provided by multidisciplinary teams of specialists including ophthalmology, pediatric oncology, pathology, and radiation oncology. Treatment options depend on tumor stage, number of tumor foci (unifocal, unilateral multifocal, or bilateral), localization and size of the tumor(s) within the eye(s), presence of vitreous seeding, potential for useful vision, extent and kind of extraocular extension, and resources available. Treatment options include enucleation; cryotherapy; laser; systemic or local ocular chemotherapy, including intra-arterial chemotherapy, combined with or followed by laser or cryotherapy; radiation therapy using episcleral plaques; and, as a last resort, external beam radiotherapy. Standard treatments for non-ocular neoplasms.
Surveillance: Individuals with an RB1 germline pathogenic variant (H1) should have eye examinations (under anesthesia in young children) every three to four weeks until age six months, every two months until age three years, every three to six months until age seven years, annually until age ten years, and then every two years in order to identify a retinoblastoma as early as possible. In individuals with unilateral retinoblastoma without an identified heterozygous germline RB1 pathogenic variant (H0*), clinical eye examination and eye ultrasound every three to six months until age seven years, then every two years. In individuals with retinomas, retinal examinations and imaging every one to two years. To detect non-ocular tumors, prompt clinical investigation of any signs and/or symptoms of malignant neoplasms.
Agents/circumstances to avoid: If possible, radiation (including x-rays, CT scans, and external beam radiation) and DNA damaging agents (tobacco, UV light) should be avoided in individuals with heritable retinoblastoma (H1) to minimize the lifetime risk of developing subsequent malignant neoplasms. It is plausible that cancer risks in these individuals may also be reduced by limiting exposure to chemotherapy.
Evaluation of relatives at risk: Molecular genetic testing if the pathogenic variant in the family is known (which can reduce the need for costly screening procedures in those family members who have not inherited the pathogenic variant [i.e., H0]) or eye examinations by an ophthalmologist experienced in the treatment of retinoblastoma if the pathogenic variant is not known for early identification of asymptomatic at-risk children.
Genetic counseling.
Heritable retinoblastoma is inherited in an autosomal dominant manner. The majority of individuals with heritable retinoblastoma represent simplex cases (i.e., the only person in the family known to be affected). Some individuals diagnosed with heritable retinoblastoma inherited an RB1 pathogenic variant from an affected parent. Each child of an individual with heritable retinoblastoma (H1) has a 50% chance of inheriting the RB1 pathogenic variant. If the RB1 pathogenic variants that have been detected in tumor tissue are not detected in DNA from leukocytes of the proband, there is an estimated 1.2% chance that the proband has germline mosaicism for one of the pathogenic variants identified in the tumor tissue and the offspring of the proband are at a 0.6% risk of inheriting a germline pathogenic variant. If one of the pathogenic variants identified in the proband's tumor is found to be mosaic in DNA from leukocytes of the proband, the level of germline involvement is uncertain. Predictive DNA testing in offspring is possible if the cancer-predisposing germline RB1 variant has been identified in the proband. If molecular genetic testing is not available or is uninformative in the proband, empiric risks to offspring based on tumor presentation and family history can be used. If the germline RB1 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Diagnosis
Guidelines for diagnosis and care of children and families affected by retinoblastoma have been published [Canadian Retinoblastoma Society 2009].
Suggestive Findings
Retinoblastoma should be suspected in children with any of the following:
- Leukocoria (white pupil)
- Strabismus
- Change in eye appearance
- Reduced visual acuity
Heritable retinoblastoma should be suspected in an individual with any of the following:
- Diagnosis of retinoblastoma, including unilateral (unifocal and multifocal) and bilateral involvement
- Retinoma
- Family history of retinoblastoma
Establishing the Diagnosis
The diagnosis of retinoblastoma is established in a proband by retinal examination with full pupillary dilatation by an ophthalmologist or optometrist. Confirmation of the diagnosis and determination of the disease extent is accomplished by examination under anesthesia. Ocular imaging can help confirm the diagnosis. Pathology is not required. Note: Biopsy may cause the tumor to spread beyond the eye, endangering the life of the individual.
The diagnosis of heritable retinoblastoma is established in a proband with retinoblastoma or retinoma and a family history of retinoblastoma. However, the majority of individuals with retinoblastoma do not have a family history of the disorder. These individuals require identification of a heterozygous germline RB1 pathogenic (or likely pathogenic) variant on molecular genetic testing (see Table 1) to determine if the retinoblastoma is heritable. Identification of an RB1 pathogenic variant in the proband allows for early diagnosis and screening for relatives at risk for retinoblastoma.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of a heterozygous RB1 variant of uncertain significance does not establish or rule out the diagnosis.
The following staging has been recommended to clarify genetic risk of a germline RB1 pathogenic variant [Mallipatna et al 2017, Soliman et al 2017a]:
- HX. Individual with unknown or insufficient evidence of a constitutional (germline) RB1 pathogenic variant
- H0. Individual who did not inherit a known familial germline RB1 pathogenic variant confirmed by molecular genetic testing
- (H0*. Individual with unilateral retinoblastoma or retinoma with no germline RB1 pathogenic variant identified on molecular genetic testing; residual risk of mosaicism is <1% on test certified to detect low-level mosaicism.)
- H1. Individual with bilateral retinoblastoma, trilateral retinoblastoma (retinoblastoma with intracranial central nervous system midline embryonic tumor), retinoblastoma and a family history of retinoblastoma, or identification of a germline RB1 pathogenic variant
Molecular genetic testing approaches to identify individuals with heritable retinoblastoma can include single-gene testing and chromosomal microarray (CMA).
Single-gene testing
- Individuals with bilateral, unilateral familial, or unilateral multifocal retinoblastoma. Sequence analysis and gene-targeted deletion/duplication analysis of RB1 are performed on peripheral blood DNA.
- Individuals with unilateral unifocal retinoblastoma and a negative family history
- If tumor tissue is not available, sequence analysis and gene-targeted deletion/duplication analysis of RB1 are performed on peripheral blood DNA.
- If tumor tissue is available, sequence analysis and gene-targeted deletion/duplication analysis of RB1 are performed on tumor DNA. If pathogenic variants are identified, DNA from blood is tested for the presence of these variants. If no pathogenic variants are identified in the tumor, methylation analysis of the RB1 promoter CpG island is performed to identify epigenetic inactivation of RB1 resulting from hypermethylation of the RB1 promoter. If no hypermethylation is identified at the promoter, DNA from tumor is tested for the amplification of MYCN, which is the cause of unilateral, non-heritable retinoblastoma in the absence of RB1 pathogenic variants in about 1.5% of individuals with isolated unilateral retinoblastoma [Rushlow et al 2013].
CMA uses oligonucleotide or SNP arrays to detect genome-wide large deletions/duplications (including RB1) that cannot be detected by sequence analysis. CMA may be considered in individuals with retinoblastoma associated with developmental delay and/or other congenital anomalies [Mitter et al 2011, Castéra et al 2013].
Table 1.
Molecular Genetic Testing Used in Heritable Retinoblastoma
Table 2.
Probability of a Proband with Retinoblastoma Having an RB1 Germline Pathogenic Variant Based on Family History and Tumor Presentation
Note: (1) If neither RB1 pathogenic variant identified in tumor tissue is found in the DNA of non-tumor cells (constitutional DNA), the affected individual has a low probability of having an RB1 germline pathogenic variant. (2) Although deep next-generation sequencing can detect low-level somatic mosaicism for a pathogenic variant, the failure to detect an RB1 pathogenic variant in constitutional DNA reduces but cannot fully eliminate the probability that the individual has an RB1 pathogenic variant in their germline.
Other testing. Molecular analysis of cell-free DNA from the aqueous humor in the anterior chamber of an eye with retinoblastoma can be used to identify the biallelic RB1 pathogenic variants without removal of the eye [Berry et al 2018, Xu et al 2021]. To date, this testing is not clinically available for exclusion of heritable predisposition to retinoblastoma.
Clinical Characteristics
Clinical Description
Retinoblastoma. The most common presenting sign is a white pupillary reflex (leukocoria). Strabismus is the second most common presenting sign and may accompany or precede leukocoria [Abramson et al 2003]. Unusual presenting signs include glaucoma, orbital cellulitis, uveitis, hyphema, or vitreous hemorrhage. Most affected children are diagnosed before age five years. Atypical manifestations are more frequent in older children.
Probands with retinoblastoma usually present in one of the following clinical settings:
- Negative family history and unilateral retinoblastoma (60% of probands)
- Negative family history and bilateral retinoblastoma (30% of probands)
- Positive family history and unilateral or bilateral retinoblastoma (~10% of probands). For H1 individuals (see Establishing the Diagnosis) with a positive family history who undergo clinical surveillance via serial retinal examinations, tumors are often identified in the first month of life.
- Chromosome deletion involving band 13q14. Up to 5% of all probands with unifocal retinoblastoma and 7.5% of all probands with multifocal retinoblastoma have a chromosome deletion of 13q14. Such chromosome abnormalities are often associated with developmental delay and birth defects [Mitter et al 2011, Castéra et al 2013].
Retinoblastoma is:
- Unilateral if only one eye is affected by retinoblastoma. About 60% of affected individuals have unilateral retinoblastoma with a mean age at diagnosis of 24 months. In individuals with unilateral retinoblastoma the tumor is usually also unifocal (i.e., only a single tumor is present). Some individuals have multifocal tumors in one eye (unilateral multifocal retinoblastoma). Intraocular seeding may mimic primary multifocal tumor growth. In most persons with unilateral retinoblastoma without a family history, the tumor is large, and it is not possible to determine if a single tumor is present.
- Bilateral if both eyes are affected by retinoblastoma. About 40% of affected individuals have bilateral retinoblastoma with a mean age at diagnosis of 15 months. In most children with bilateral tumors, both eyes are affected at the time of initial diagnosis. In individuals with bilateral retinoblastoma, both eyes may show multiple tumors. Some children who are initially diagnosed with unilateral retinoblastoma later develop a tumor in the contralateral unaffected eye.
- Trilateral if bilateral (or, rarely, unilateral) retinoblastoma and a pinealoblastoma develop (see Pinealoblastomas).
Retinoma and associated eye lesions. Benign retinal tumors (i.e., retinoma) that have undergone spontaneous growth arrest may present within retinal scars [Dimaras et al 2008]. Calcified phthisic eyes may result from spontaneous regression of retinoblastoma associated with vascular occlusion [Valverde et al 2002]. The spectrum of RB1 pathogenic variants in individuals with retinoma and a family history of retinoblastoma and individuals who had retinoma in one eye and either retinoma or retinoblastoma in the other eye appears to be indistinct from that of individuals with bilateral retinoblastoma [Abouzeid et al 2009].
Pinealoblastomas occur in "retina-like" tissue in the pineal gland of the brain. Concurrence of pinealoblastomas or primitive neuroectodermal tumors and retinoblastoma is referred to as trilateral retinoblastoma. Pinealoblastoma is rare and usually fatal, unlike retinoblastoma of the eye, which is generally curable [de Jong et al 2014].
Other tumors. There is an increased risk for other specific extraocular subsequent malignant neoplasms (collectively called second primary tumors). Most common subsequent malignant neoplasms are osteosarcoma, soft tissue sarcoma (mostly leiomyosarcoma and rhabdomyosarcoma), or melanoma [Kleinerman et al 2007, Marees et al 2008, Kleinerman et al 2012]. These tumors usually manifest in adolescence or adulthood. The incidence of subsequent malignant neoplasms is more than 50% in individuals with retinoblastoma who received external beam radiation therapy [Wong et al 1997]. Survivors of heritable retinoblastoma who were not exposed to high-dose radiotherapy have a lower, but still high, lifetime risk of subsequent malignant neoplasms [Dommering et al 2012b, Kleinerman et al 2012, MacCarthy et al 2013, Temming et al 2015, Chaussade et al 2019, Kleinerman et al 2019, Ketteler et al 2020].
Genotype-Phenotype Correlations
In the majority of families with heritable retinoblastoma, all members who inherited the germline pathogenic variant develop multiple tumors in both eyes. It is not unusual to find, however, that the founder (i.e., the first person in the family to have retinoblastoma) has only unilateral retinoblastoma. These families may have RB1 null alleles that are altered by frameshift or nonsense variants, or the founder may be mosaic for the pathogenic RB1 allele. With few exceptions, RB1 null alleles show complete penetrance (>99%) [Taylor et al 2007, Dommering et al 2014, Frenkel et al 2016].
Fewer than 10% of families show a "low-penetrance" phenotype with reduced expressivity (i.e., increased prevalence of unilateral retinoblastoma) and reduced penetrance (i.e., ≤25%). This low-penetrance phenotype is usually associated with RB1 in-frame, missense, or distinct splice site variants, certain indel variants in exon 1, or pathogenic variants in the promoter region.
A third category of families show differential penetrance depending on the parental origin of the pathogenic allele (parent-of-origin effect) [Klutz et al 2002, Eloy et al 2016, Imperatore et al 2018].
Cytogenetically visible deletions of 13q14 that include genes adjacent to RB1 may cause developmental delay [Castéra et al 2013] and mild-to-moderate facial dysmorphism. Large deletions of 13q14 show reduced expressivity, and individuals may have unilateral retinoblastoma or absence of retinoblastoma [Mitter et al 2011]. Contiguous loss of MED4, which is located centromeric to RB1, explains reduced expressivity in individuals with large deletions that include both RB1 and MED4 [Dehainault et al 2014].
Penetrance
Nomenclature
Glioma retinae is a historical name for retinoblastoma.
Prevalence
The incidence of retinoblastoma is estimated at between 1:15,000 and 1:20,000 live births [Moll et al 1997, Seregard et al 2004, Nummi & Kivelä 2021].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in RB1.
Sporadic tumors including bladder urothelial cancer, endometrial cancer, sarcoma, squamous cell cancer of the lung, and lung adenocarcinoma (portal.gdc.cancer.gov) occurring as single tumors in the absence of any other findings of heritable retinoblastoma frequently contain a somatic variant in RB1 that is not present in the germline. In these circumstances predisposition to these tumors is not heritable.
Differential Diagnosis
Several hereditary ocular conditions of childhood can clinically simulate retinoblastoma:
- NDP-related persistent fetal vasculature and NDP-related Coats disease (See NDP-Related Retinopathies.)
- Norrie disease (See NDP-Related Retinopathies.)
- Familial exudative vitreoretinopathy (See Phenotypic Series: Exudative Vitreoretinopathy.)
Ocular infestation by Toxocara canis can also clinically simulate retinoblastoma.
Management
Guidelines for retinoblastoma care have been developed (see Canadian Retinoblastoma Society [2009] and Kenya National Retinoblastoma Strategy Best Practice Guidelines).
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with retinoblastoma, the following evaluations (if not performed as part of the evaluation that led to the diagnosis) are recommended:
- Prior to planning therapy, the extent of the tumor within and outside the eye should be determined. Each affected eye is assigned a cancer stage, depending on the extent of disease and the risk that the cancer has spread outside the eye [Mallipatna et al 2017]. Extent of the tumor is estimated by clinical examination under anesthesia and ultrasound or MRI, particularly focusing on the tumor-optic nerve relationship. Brain MRI is also useful to evaluate for a pinealoblastoma, indicating trilateral retinoblastoma. CT examination is NOT recommended because of the potentially increased risk for subsequent malignant neoplasms due to radiation exposure.
- For very large tumors with risk factors for extraocular disease, bone marrow aspiration and examination of cerebrospinal fluid may also be performed at diagnosis, or when pathologic examination of the enucleated eye reveals optic nerve invasion or significant risks for extraocular extension.
- If retinoblastoma has spread outside the eye, the stage of cancer will be evaluated to determine the most appropriate care for the child.
- In individuals with a family history of retinoblastoma, and in uncommon circumstances in which the child presents with strabismus or poor vision, the retinal tumors may be small and detected by optical coherence tomography [Soliman et al 2017b].
- Consultation with a medical geneticist, certified genetic counselor, or certified advanced genetic nurse to inform affected individuals and their families about the nature, mode of inheritance, and implications of retinoblastoma can facilitate medical and personal decision making.
Treatment of Manifestations
Goals of treatment in order of priority are preservation of life and then sight. As optimal treatment may be complex, specialists skilled in the treatment of retinoblastoma from various fields including ophthalmology, pediatric oncology, pathology, and radiation oncology collaborate to deliver optimized care.
In addition to eye and tumor stage, choice of treatment depends on many factors, including the number of tumor foci (unifocal, unilateral multifocal, or bilateral), localization and size of the tumor(s) within the eye(s), presence of vitreous seeding, potential for useful vision, extent and kind of extraocular extension, and resources available.
Treatment options for the eye include enucleation; cryotherapy; laser; systemic or local ocular chemotherapy, including intra-arterial chemotherapy, combined with or followed by laser or cryotherapy; radiation therapy using episcleral plaques; and, as a last resort, external beam radiotherapy.
If possible, radiation (including x-ray, CT scan, and external beam radiation) is avoided to minimize the lifetime risk of developing late-onset subsequent malignant neoplasms. Such tests may be used when critical in essential health care.
Although subsequent malignant neoplasms share a molecular feature (RB1 inactivation), their treatment is presently not distinct from similar types of cancer that arose without heritable predisposition.
Surveillance
Guidelines for clinical screening for children at risk have been published [Skalet et al 2018]. Further information regarding medical surveillance for those who have had or are at risk of developing retinoblastoma is available in the guidelines for retinoblastoma care. Children who have undergone successful treatment require frequent follow-up examination for early detection of newly arising intraocular tumors, as indicated in the guidelines.
Table 3.
Retinoblastoma: Recommended Surveillance for Individuals at Risk and Affected Individuals
Agents/Circumstances to Avoid
It has been suggested by Fletcher et al [2004] that cancer risks in survivors of heritable retinoblastoma may be reduced by limiting exposure to DNA-damaging agents (radiotherapy, tobacco, and UV light) [Temming et al 2017]. It is plausible that cancer risks in these individuals may also be reduced by limiting exposure to chemotherapy.
Evaluation of Relatives at Risk
The American Society of Clinical Oncologists identifies heritable retinoblastoma as a Group 1 disorder – a hereditary syndrome for which genetic testing is considered part of the standard management for at-risk family members [American Society of Clinical Oncology 2003, Robson et al 2010]. It is appropriate to evaluate apparently asymptomatic at-risk relatives of an affected individual in order to identify as early as possible those who would benefit from eye examination by an experienced ophthalmologist and allow for early identification and treatment of a retinoblastoma and/or subsequent non-ocular malignancies. Early diagnosis and treatment of retinoblastoma and RB1-related subsequent malignant neoplasms can reduce morbidity and increase longevity. Evaluations can include:
- Molecular genetic testing if the pathogenic variant in the family is known, which reduces the need for costly screening procedures in those at-risk family members who have not inherited the pathogenic variant [Noorani et al 1996, Richter et al 2003, Skalet et al 2018].
- Eye examinations by an ophthalmologist experienced in the treatment of retinoblastoma starting directly after birth (see Table 3). Young or uncooperative children may require examination under anesthesia.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
NCT04428879. This single-site, single-arm, non-randomized, dose-escalation Phase I toxicity clinical trial will assess primarily the safety and secondarily the efficacy of episcleral topotecan in individuals with active de novo or residual or recurrent intraocular retinoblastoma in at least one eye following completion of first-line therapy.
NCT04156347. This single-arm, non-randomized, dose-escalation Phase I clinical trial will assess primarily the safety and secondarily the efficacy of episcleral topotecan in individuals with active de novo or recurrent intraocular retinoblastoma in at least one eye following completion of first-line therapy.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Heritable retinoblastoma is an autosomal dominant disorder.
Risk to Family Members
Parents of a proband
- The majority of individuals with heritable retinoblastoma represent simplex cases (i.e., the only person in the family known to be affected).
- Most individuals with bilateral tumors who represent simplex cases have heritable retinoblastoma as the result of a de novo RB1 pathogenic variant.
- Approximately 5% of individuals with bilateral tumors who represent simplex cases have the disorder as the result of an RB1 pathogenic variant inherited from an unaffected parent; the transmitting parent may be mosaic for the RB1 pathogenic variant found in the bilaterally affected child, or they may be heterozygous for a "reduced-penetrance" pathogenic variant [Rushlow et al 2009, Skalet et al 2018].
- Some individuals diagnosed with heritable retinoblastoma inherited an RB1 pathogenic variant from an affected parent.
- If a germline RB1 pathogenic variant has been identified in the proband and the proband appears to be the only affected family member, molecular genetic testing is recommended for the parents of the proband to evaluate their genetic status and inform recurrence risk assessment.* If the RB1 pathogenic variant in the proband is not known, recommendations for the evaluation of parents include examination by an ophthalmologist knowledgeable about retinoblastoma, retinoma, and retinoblastoma-associated eye lesions.* The possibility that a parent has an RB1 pathogenic variant can be excluded if the proband with retinoblastoma has the disorder as the result of a somatic mosaic RB1 pathogenic variant (i.e., a pathogenic variant that occurred in the proband during embryonic development). With methods such as allele-specific PCR [Rushlow et al 2009] or next-generation sequencing [Chen et al 2014], mosaicism is evident in 5.5% of probands with bilateral retinoblastoma and 3.8% of probands with unilateral retinoblastoma.
- If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism.* Note: Testing parents using peripheral blood leukocyte-derived DNA may not detect all instances of somatic mosaicism. Molecular genetic tests sensitive enough to detect low-level mosaicism, such as high-coverage next-generation sequencing or allele-specific PCR, can be considered.* If the parent is the individual in whom the pathogenic variant first occurred, the parent may have somatic and germline mosaicism for the pathogenic variant and have fewer (unilateral) or no retinoblastomas.
- The family history of some individuals diagnosed with heritable retinoblastoma may appear to be negative because of failure to recognize a retinoma in a family member, low-level mosaicism, or reduced penetrance. Therefore, a negative family history may require both clinical evaluation and molecular genetic testing demonstrating that neither parent is heterozygous for the RB1 pathogenic variant identified in the proband.
Sibs of a proband. The risk to sibs of a proband depends on the phenotype and the genetic status of the parents:
- Risk to sibs based on the phenotype of the parents:
- If a parent of the proband and the proband have bilateral retinoblastoma, the risk to the sibs is 50%. In rare families with "familial low-penetrance retinoblastoma," the risk of tumor development in a sib with the germline pathogenic variant is reduced (see Genotype-Phenotype Correlations).
- If the parents are clinically unaffected, the risk to the sibs appears to be low (i.e., 1%-2%; see Table 4) but is still increased because of the possibility of reduced penetrance in a heterozygous parent or parental germline mosaicism.
- Risk to sibs based on the genetic status of the parents:
- If a parent is heterozygous for the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50% and testing of sibs for the RB1 pathogenic variant is warranted. In rare families with "familial low-penetrance retinoblastoma," the risk of tumor development in a sib with the germline pathogenic variant is reduced (see Genotype-Phenotype Correlations).
- If the RB1 pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is low (<5%) but greater than that of the general population because of the possibility of parental germline mosaicism. Sibs can be tested for the RB1 pathogenic variant identified in the proband. If the known familial RB1 pathogenic variant is not identified, the sib is considered "H0" (i.e., an individual who did not inherit a known familial germline RB1 pathogenic variant confirmed by molecular genetic testing) [Mallipatna et al 2017, Soliman et al 2017a, Skalet et al 2018].
- If the proband clearly shows mosaicism for an RB1 cancer-predisposing variant in non-cancer cells such as leukocyte DNA, it is assumed that the pathogenic variant arose as a postzygotic event and that neither parent has an RB1 germline pathogenic variant. The risk to the sibs is not increased and thus the testing of sibs for the RB1 pathogenic variant identified in the proband is not warranted.
- If molecular genetic testing is not available or is uninformative, empiric risks based on tumor presentation (e.g., unifocal or multifocal) and family history can be used (see Table 4) [Skalet et al 2018]. The low, but not negligible, risk to sibs of a proband with a negative family history presumably reflects the presence of either a germline RB1 pathogenic variant with reduced penetrance in one parent or somatic mosaicism (that includes the germline) for an RB1 pathogenic variant in one parent.
- If a parent has a cytogenetically detectable balanced chromosome 13 translocation or rearrangement, the sibs are at increased risk of inheriting an unbalanced chromosome rearrangement.
Table 4.
Pretest Empiric Risks for Development of Retinoblastoma in Sibs of a Proband Based on Family History and Retinoblastoma Presentation in the Proband
Offspring of a proband. Each child of an individual with heritable retinoblastoma (H1) has a 50% chance of inheriting the RB1 pathogenic variant.
- If the RB1 pathogenic variants that have been detected in tumor tissue are not detected in DNA from leukocytes of the proband, there is an estimated 1.2% chance that the proband has germline mosaicism for one of the pathogenic variants identified in the tumor tissue. The offspring of the proband are at a 0.6% risk of inheriting a germline pathogenic variant [Richter et al 2003]. Molecular genetic testing in offspring should include analysis for both of the pathogenic variants identified in the proband's tumor tissue.
- If one of the pathogenic variants identified in the proband's tumor is found to be mosaic in DNA from leukocytes of the proband, the level of germline involvement is uncertain. All offspring should have molecular genetic testing for the pathogenic variant identified in leukocyte DNA of the proband.
- Predictive DNA testing in offspring is possible if the cancer-predisposing RB1 variant has been identified in the proband.
- If molecular genetic testing is not available or is uninformative in the proband, empiric risks based on tumor presentation and family history can be used (see Table 5) [Skalet et al 2018].
Table 5.
Pretest Empiric Risks for Development of Retinoblastoma in Offspring of a Proband Based on Family History and Retinoblastoma Presentation in the Proband
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected, the parent's family members may be at risk.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Predictive testing (i.e., testing of asymptomatic at-risk individuals)
- Predictive testing for at-risk asymptomatic family members requires prior identification of the germline RB1 pathogenic variant in the family.
- Predictive molecular genetic testing of young, at-risk family members is appropriate for guiding medical management (see Management, Evaluation of Relatives at Risk).
- Potential consequences of such testing – including but not limited to evaluation arrangements for individuals with a positive test result – as well as the capabilities and limitations of predictive testing should be discussed in the context of formal genetic counseling prior to testing.
Genetic cancer risk assessment and counseling. For a comprehensive description of the medical, psychosocial, and ethical ramifications of identifying at-risk individuals through cancer risk assessment with or without molecular genetic testing, see Cancer Genetics Risk Assessment and Counseling – for health professionals (part of PDQ®, National Cancer Institute).
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk [Dommering et al 2012a].
Prenatal Testing and Preimplantation Genetic Testing
If the germline RB1 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
When there is a family history of retinoblastoma, various options are available to optimize management of an at-risk pregnancy [Canadian Retinoblastoma Society 2009, Soliman et al 2016]:
- If an RB1 pathogenic variant is identified in the fetus, ultrasound examination may be used to identify medium-sized intraocular tumors. If tumors are present, preterm delivery to enable early treatment may be considered [Sahgal et al 2006]. Even if no tumors are visible on obstetric ultrasound, delivery of the fetus at 36 weeks' gestation may be recommended, as 30% of infants with an RB1 pathogenic variant will have a tiny vision-threatening tumor [Soliman et al 2016, Soliman et al 2018].
- If the RB1 pathogenic variant has not been identified in an affected family member, prenatal ultrasound or MRI may reveal a moderately large retinoblastoma in the eye of an affected fetus; however, these tests are not sensitive enough to detect small retinoblastoma tumors.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal and preimplantation genetic testing. While most health care professionals would consider use of prenatal and preimplantation genetic testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Childhood Eye Cancer Trust (CHECT)United KingdomPhone: +44 020 7377 5578Email: info@chect.org.uk
- MedlinePlus
- NCBI Genes and Disease
- World Eye Cancer Hope (WE C Hope)
- American Childhood Cancer OrganizationPhone: 855-858-2226
- National Cancer Institute (NCI)Phone: 800-4-CANCEREmail: NCIinfo@nih.gov
- National Federation of the BlindPhone: 410-659-9314Email: nfb@nfb.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Retinoblastoma: Genes and Databases
Table B.
OMIM Entries for Retinoblastoma (View All in OMIM)
Molecular Pathogenesis
RB1 encodes a ubiquitously expressed nuclear protein that is involved in cell cycle regulation (G1 to S transition). The RB protein is phosphorylated by members of the cyclin-dependent kinase (CDK) system prior to the entry into S phase. On phosphorylation, the binding activity of the pocket domain is lost, resulting in the release of cellular proteins. For a review see Dick & Rubin [2013], Dimaras et al [2015], and Dyson [2016].
The majority of RB1 pathogenic variants result in a premature termination codon, usually through single-base substitutions, frameshift variants, or out-of-frame exon skipping caused by splice site variants. Pathogenic variants in RB1 lead to the expression of proteins that have lost cell cycle-regulating functions. Retention of partial activity has been observed in proteins resulting from pathogenic variants that are associated with low-penetrance retinoblastoma [Bremner et al 1997, Otterson et al 1997, Sánchez-Sánchez et al 2007].
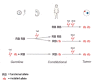
Mechanism of disease causation. Loss of function. Retinoblastoma tumor development starts when a developing retinal cell in the cone photoreceptor lineage loses both RB1 alleles (see Figure 1) [Rushlow et al 2013, Xu et al 2014].
Chapter Notes
Author Notes
Contact Dr Dietmar Lohmann (moc.cam@amotsalboniter) to inquire about review of RB1 variants of uncertain significance.
Author History
Norbert Bornfeld, MD; University of Essen (2000-2004)
Brenda L Gallie, MD (2004-present)
Bernhard Horsthemke, PhD; University of Essen (2000-2004)
Dietmar R Lohmann, MD (2000-present)
Eberhard Passarge, MD; University of Essen (2000-2004)
Revision History
- 21 September 2023 (sw) Comprehensive update posted live
- 21 November 2018 (sw) Comprehensive update posted live
- 19 November 2015 (me) Comprehensive update posted live
- 28 March 2013 (me) Comprehensive update posted live
- 10 June 2010 (me) Comprehensive update posted live
- 7 May 2007 (me) Comprehensive update posted live
- 28 December 2004 (me) Comprehensive update posted live
- 21 January 2003 (me) Comprehensive update posted live
- 18 July 2000 (me) Review posted live
- 21 January 1999 (dl) Original submission
References
Published Guidelines / Consensus Statements
- American Society of Clinical Oncology. Policy statement update: genetic testing for cancer susceptibility. Available online. 2010. Accessed 8-29-23.
- American Society of Clinical Oncology. Statement on genetic testing for cancer susceptibility. 2003.
- Canadian Retinoblastoma Society. National Retinoblastoma Strategy Canadian Guidelines for Care: Stratégie thérapeutique du rétinoblastome guide clinique canadien. Can J Ophthalmol. 2009;44:S1-88. [PubMed]
- Kenya Ministry of Health Ophthalmic Services Unit. Kenya national retinoblastoma strategy best practice guidelines 2014. Available online. 2014. Accessed 8-31-23.
Literature Cited
- Abouzeid H, Schorderet DF, Balmer A, Munier FL. Germline mutations in retinoma patients: relevance to low-penetrance and low-expressivity molecular basis. Mol Vis. 2009;15:771-7. [PMC free article: PMC2671583] [PubMed: 19390654]
- Abramson DH, Beaverson K, Sangani P, Vora RA, Lee TC, Hochberg HM, Kirszrot J, Ranjithan M. Screening for retinoblastoma: presenting signs as prognosticators of patient and ocular survival. Pediatrics. 2003;112:1248-55. [PubMed: 14654593]
- American Society of Clinical Oncology. Statement on genetic testing for cancer susceptibility. 2003. [PubMed: 12692171]
- Berry JL, Xu L, Kooi I, Murphree AL, Prabakar RK, Reid M, Stachelek K, Le BHA, Welter L, Reiser BJ, Chevez-Barrios P, Jubran R, Lee TC, Kim JW, Kuhn P, Cobrinik D, Hicks J. Genomic cfDNA analysis of aqueous humor in retinoblastoma predicts eye salvage: the surrogate tumor biopsy for retinoblastoma. Mol Cancer Res. 2018;16:1701-12. [PMC free article: PMC6214755] [PubMed: 30061186]
- Bremner R, Du DC, Connolly-Wilson MJ, Bridge P, Ahmad KF, Mostachfi H, Rushlow D, Dunn JM, Gallie BL. Deletion of RB exons 24 and 25 causes low-penetrance retinoblastoma. Am J Hum Genet. 1997;61:556-70. [PMC free article: PMC1715941] [PubMed: 9326321]
- Canadian Retinoblastoma Society. National Retinoblastoma Strategy Canadian Guidelines for Care: Stratégie thérapeutique du rétinoblastome guide clinique canadien. Can J Ophthalmol. 2009;44:S1-88. [PubMed: 20237571]
- Castéra L, Dehainault C, Michaux D, Lumbroso-Le Rouic L, Aerts I, Doz F, Pelet A, Couturier J, Stoppa-Lyonnet D, Gauthier-Villars M, Houdayer C. Fine mapping of whole RB1 gene deletions in retinoblastoma patients confirms PCDH8 as a candidate gene for psychomotor delay. Eur J Hum Genet. 2013;21:460-4. [PMC free article: PMC3598316] [PubMed: 22909775]
- Chaussade A, Millot G, Wells C, Brisse H, Laé M, Savignoni A, Desjardins L, Dendale R, Doz F, Aerts I, Jimenez I, Cassoux N, Stoppa Lyonnet D, Gauthier Villars M, Houdayer C. Correlation between RB1germline mutations and second primary malignancies in hereditary retinoblastoma patients treated with external beam radiotherapy. Eur J Med Genet. 2019;62:217-23. [PubMed: 30031154]
- Chen Z, Moran K, Richards-Yutz J, Toorens E, Gerhart D, Ganguly T, Shields CL, Ganguly A. Enhanced sensitivity for detection of low-level germline mosaic RB1 mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum Mutat. 2014;35:384-91. [PMC free article: PMC4112364] [PubMed: 24282159]
- Davies HR, Broad KD, Onadim Z, Price EA, Zou X, Sheriff I, Karaa EK, Scheimberg I, Reddy MA, Sagoo MS, Ohnuma SI, Nik-Zainal S. Whole-genome sequencing of retinoblastoma reveals the diversity of rearrangements disrupting RB1 and uncovers a treatment-related mutational signature. Cancers (Basel). 2021;13:754. [PMC free article: PMC7918943] [PubMed: 33670346]
- Dehainault C, Garancher A, Castéra L, Cassoux N, Aerts I, Doz F, Desjardins L, Lumbroso L, Montes de Oca R, Almouzni G, Stoppa-Lyonnet D, Pouponnot C, Gauthier-Villars M, Houdayer C. The survival gene MED4 explains low penetrance retinoblastoma in patients with large RB1 deletion. Hum Mol Genet. 2014;23:5243-50. [PubMed: 24858910]
- de Jong MC, Kors WA, de Graaf P, Castelijns JA, Kivela T, Moll AC. Trilateral retinoblastoma: a systematic review and meta-analysis. Lancet Oncol. 2014;15:1157-67. [PubMed: 25126964]
- Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14:297-306. [PMC free article: PMC4754300] [PubMed: 23594950]
- Dimaras H, Corson D, Cobrinik D, White A, Zhao J, Munier FL, Abramson DH, Shields CL, Chantada GL, Njuguna F, Gallie BL. Retinoblastoma. Nat Rev Dis Primers. 2015;1:15021. [PMC free article: PMC5744255] [PubMed: 27189421]
- Dimaras H, Khetan V, Halliday W, Orlic M, Prigoda NL, Piovesan B, Marrano P, Corson TW, Eagle RC Jr, Squire JA, Gallie BL. Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008;17:1363-72. [PubMed: 18211953]
- Dommering CJ, Garvelink MM, Moll AC, van Dijk J, Imhof SM, Meijers-Heijboer H, Henneman L. Reproductive behavior of individuals with increased risk of having a child with retinoblastoma. Clin Genet. 2012a;81:216-23. [PubMed: 21954974]
- Dommering CJ, Marees T, van der Hout AH, Imhof SM, Meijers-Heijboer H, Ringens PJ, van Leeuwen FE, Moll AC. RB1 mutations and second primary malignancies after hereditary retinoblastoma. Fam Cancer. 2012b;11:225-33. [PMC free article: PMC3365233] [PubMed: 22205104]
- Dommering CJ, Mol BM, Moll AC, Burton M, Cloos J, Dorsman JC, Meijers-Heijboer H, van der Hout AH. RB1 mutation spectrum in a comprehensive nationwide cohort of retinoblastoma patients. J Med Genet. 2014;51:366–74. [PubMed: 24688104]
- Draper GJ, Sanders BM, Brownbill PA, Hawkins MM. Patterns of risk of hereditary retinoblastoma and applications to genetic counselling. Br J Cancer. 1992;66:211-9. [PMC free article: PMC1977909] [PubMed: 1637670]
- Dyson NJ. RB1: a prototype tumor suppressor and an enigma. Genes Dev. 2016;30:1492–502. [PMC free article: PMC4949322] [PubMed: 27401552]
- Eloy P, Dehainault C, Sefta M, Aerts I, Doz F, Cassoux N, Lumbroso le Rouic L, Stoppa-Lyonnet D, Radvanyi F, Millot GA, Gauthier-Villars M, Houdayer C. A parent-of-origin effect impacts the phenotype in low penetrance retinoblastoma families segregating the c.1981C>T/p.Arg661Trp mutation of RB1. PLoS Genet. 2016;12:e1005888. [PMC free article: PMC4771840] [PubMed: 26925970]
- Fletcher O, Easton D, Anderson K, Gilham C, Jay M, Peto J. Lifetime risks of common cancers among retinoblastoma survivors. J Natl Cancer Inst. 2004;96:357-63. [PubMed: 14996857]
- Frenkel S, Zloto O, Sagi M, Fraenkel A, Pe'er J. Genotype-phenotype correlation in the presentation of retinoblastoma among 149 patients. Exp Eye Res. 2016;146:313–7. [PubMed: 27068507]
- Imperatore V, Pinto AM, Gelli E, Trevisson E, Morbidoni V, Frullanti E, Hadjistilianou T, De Francesco S, Toti P, Gusson E, Roversi G, Accogli A, Capra V, Mencarelli MA, Renieri A, Ariani F. Parent-of-origin effect of hypomorphic pathogenic variants and somatic mosaicism impact on phenotypic expression of retinoblastoma. Eur J Hum Genet. 2018;26:1026–37. [PMC free article: PMC6018773] [PubMed: 29662154]
- Ketteler P, Hülsenbeck I, Frank M, Schmidt B, Jöckel KH, Lohmann DR. The impact of RB1 genotype on incidence of second tumours in heritable retinoblastoma. Eur J Cancer. 2020;133:47-55. [PubMed: 32434110]
- Kleinerman RA, Tucker MA, Abramson DH, Seddon JM, Tarone RE, Fraumeni JF Jr. Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst. 2007;99:24-31. [PubMed: 17202110]
- Kleinerman RA, Tucker MA, Sigel BS, Abramson DH, Seddon JM, Morton LM. Patterns of cause-specific mortality among 2053 survivors of retinoblastoma, 1914-2016. J Natl Cancer Inst. 2019;111:961-9. [PMC free article: PMC6748807] [PubMed: 30698734]
- Kleinerman RA, Yu CL, Little MP, Li Y, Abramson D, Seddon J, Tucker MA. Variation of second cancer risk by family history of retinoblastoma among long-term survivors. J Clin Oncol. 2012;30:950-7. [PMC free article: PMC3341108] [PubMed: 22355046]
- Klutz M, Brockmann D, Lohmann D. A parent-of-origin effect in two families with retinoblastoma is associated with a distinct splice mutation in the RB1 gene. Am J Hum Genet. 2002;71:174–9. [PMC free article: PMC384976] [PubMed: 12016586]
- MacCarthy A, Bayne AM, Brownbill PA, Bunch KJ, Diggens NL, Draper GJ, Hawkins MM, Jenkinson HC, Kingston JE, Stiller CA, Vincent TJ, Murphy MF. Second and subsequent tumours among 1927 retinoblastoma patients diagnosed in Britain 1951-2004. Br J Cancer. 2013;108:2455-63. [PMC free article: PMC3694232] [PubMed: 23674091]
- Mallipatna A, Gallie BL, Chévez-Barrios P, Lumbroso-Le Rouic L, Chantada GL, Doz F, et al. Retinoblastoma. In: Amin MB, Edge SB, Greene FL, eds. AJCC Cancer Staging Manual. 8 ed. New York, NY: Springer; 2017: 819-31.
- Marees T, Moll AC, Imhof SM, de Boer MR, Ringens PJ, van Leeuwen FE. Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up. J Natl Cancer Inst. 2008;100:1771-9. [PubMed: 19066271]
- Mitter D, Ullmann R, Muradyan A, Klein-Hitpass L, Kanber D, Ounap K, Kaulisch M, Lohmann D. Genotype-phenotype correlations in patients with retinoblastoma and interstitial 13q deletions. Eur J Hum Genet. 2011;19:947-58. [PMC free article: PMC3179359] [PubMed: 21505449]
- Moll AC, Kuik DJ, Bouter LM, Den Otter W, Bezemer PD, Koten JW, Imhof SM, Kuyt BP, Tan KE. Incidence and survival of retinoblastoma in The Netherlands: a register based study 1862-1995. Br J Ophthalmol. 1997;81:559-62. [PMC free article: PMC1722238] [PubMed: 9290369]
- Musarella MA, Gallie BL. A simplified scheme for genetic counseling in retinoblastoma. J Pediatr Ophthalmol Strabismus. 1987;24:124-5. [PubMed: 3598831]
- Noorani HZ, Khan HN, Gallie BL, Detsky AS. Cost comparison of molecular versus conventional screening of relatives at risk for retinoblastoma. Am J Hum Genet. 1996;59:301-7. [PMC free article: PMC1914748] [PubMed: 8755916]
- Nummi K, Kivelä TT. Retinoblastoma in Finland, 1964-2014: incidence and survival. Br J Ophthalmol. 2021;105:63-9. [PubMed: 32217545]
- Otterson GA, Chen WD, Coxon AB, Khleif SN, Kaye FJ. Incomplete penetrance of familial retinoblastoma linked to germ-line mutations that result in partial loss of RB function. Proc Natl Acad Sci U S A. 1997;94:12036-40. [PMC free article: PMC23695] [PubMed: 9342358]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Richter S, Vandezande K, Chen N, Zhang K, Sutherland J, Anderson J, Han L, Panton R, Branco P, Gallie B. Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet. 2003;72:253-69. [PMC free article: PMC379221] [PubMed: 12541220]
- Robson ME, Storm CD, Weitzel J, Wollins DS, Offit K; American Society of Clinical Oncology. American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol. 2010;28:893-901. [PubMed: 20065170]
- Rushlow D, Piovesan B, Zhang K, Prigoda-Lee NL, Marchong MN, Clark RD, Gallie BL. Detection of mosaic RB1 mutations in families with retinoblastoma. Hum Mutat. 2009;30:842-51. [PubMed: 19280657]
- Rushlow DE, Mol BM, Kennett JY, Yee S, Pajovic S, Thériault BL, Prigoda-Lee NL, Spencer C, Dimaras H, Corson TW, Pang R, Massey C, Godbout R, Jiang Z, Zacksenhaus E, Paton K, Moll AC, Houdayer C, Raizis A, Halliday W, Lam WL, Boutros PC, Lohmann D, Dorsman JC, Gallie BL. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol. 2013;14:327-34. [PubMed: 23498719]
- Sahgal A, Millar BA, Michaels H, Jaywant S, Chan HS, Heon E, Gallie B, Laperriere N. Focal stereotactic external beam radiotherapy as a vision-sparing method for the treatment of peripapillary and perimacular retinoblastoma: preliminary results. Clin Oncol (R Coll Radiol). 2006;18:628-34. [PubMed: 17051954]
- Sánchez-Sánchez F, Ramírez-Castillejo C, Weekes DB, Beneyto M, Prieto F, Nájera C, Mittnacht S. Attenuation of disease phenotype through alternative translation initiation in low-penetrance retinoblastoma. Hum Mutat. 2007;28:159–67. [PubMed: 16988938]
- Seregard S, Lundell G, Svedberg H, Kivelä T. Incidence of retinoblastoma from 1958 to 1998 in Northern Europe: advantages of birth cohort analysis. Ophthalmology. 2004;111:1228-32. [PubMed: 15177976]
- Singh HP, Shayler DWH, Fernandez GE, Thornton ME, Craft CM, Grubbs BH, Cobrinik D. An immature, dedifferentiated, and lineage-deconstrained cone precursor origin of N-Myc-initiated retinoblastoma. Proc Natl Acad Sci U S A. 2022;119:e2200721119. [PMC free article: PMC9282279] [PubMed: 35867756]
- Skalet AH, Gombos DS, Gallie BL, Kim JW, Shields CL, Marr BP, Plon SE, Chévez-Barrios P. Screening children at risk for retinoblastoma: consensus report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology. 2018;125:453-8. [PubMed: 29056300]
- Soliman S, Kletke S, Roelofs K, VandenHoven C, McKeen L, Gallie B. Precision laser therapy for retinoblastoma. Expert Rev Ophthalmol. 2018;13:149-59.
- Soliman SE, Dimaras H, Khetan V, Gardiner JA, Chan HS, Héon E, Gallie BL. Prenatal versus postnatal screening for familial retinoblastoma. Ophthalmology. 2016;123:2610-7. [PubMed: 27712844]
- Soliman SE, Racher H, Zhang C, MacDonald H, Gallie BL. Genetics and molecular diagnostics in retinoblastoma--an update. Asia Pac J Ophthalmol (Phila). 2017a;6:197-207. [PubMed: 28399338]
- Soliman SE, VandenHoven C, MacKeen LD, Heon E, Gallie BL. Optical coherence tomography-guided decisions in retinoblastoma management. Ophthalmology. 2017b;124:859-72. [PubMed: 28318638]
- Taylor M, Dehainault C, Desjardins L, Doz F, Levy C, Sastre X, Couturier J, Stoppa-Lyonnet D, Houdayer C, Gauthier-Villars M. Genotype–phenotype correlations in hereditary familial retinoblastoma. Hum Mutat. 2007;28:284–93. [PubMed: 17096365]
- Temming P, Arendt M, Viehmann A, Eisele L, Le Guin CH, Schündeln MM, Biewald E, Astrahantseff K, Wieland R, Bornfeld N, Sauerwein W, Eggert A, Jöckel KH, Lohmann DR. Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatr Blood Cancer. 2017;64:71-80. [PubMed: 27567086]
- Temming P, Viehmann A, Arendt M, Eisele L, Spix C, Bornfeld N, Sauerwein W, Jöckel KH, Lohmann DR. Pediatric second primary malignancies after retinoblastoma treatment. Pediatr Blood Cancer. 2015;62:1799-804. [PubMed: 25970657]
- Temming P, Viehmann A, Biewald E, Lohmann DR. Sporadic unilateral retinoblastoma or first sign of bilateral disease? Br J Ophthalmol. 2013;97:475-80. [PubMed: 23355526]
- Valverde K, Pandya J, Heon E, Goh TS, Gallie BL, Chan HS. Retinoblastoma with central retinal artery thrombosis that mimics extraocular disease. Med Pediatr Oncol. 2002;38:277-9. [PubMed: 11920797]
- Wong FL, Boice JD Jr, Abramson DH, Tarone RE, Kleinerman RA, Stovall M, Goldman MB, Seddon JM, Tarbell N, Fraumeni JF Jr, Li FP. Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA. 1997;278:1262-7. [PubMed: 9333268]
- Xu L, Kim ME, Polski A, Prabakar RK, Shen L, Peng CC, Reid MW, Chevez-Barrios P, Kim JW, Shah R, Jubran R, Kuhn P, Cobrinik D, Biegel JA, Gai X, Hicks J, Berry JL. Establishing the clinical utility of ctDNA analysis for diagnosis, prognosis, and treatment monitoring of retinoblastoma: the aqueous humor liquid biopsy. Cancers (Basel). 2021;13:1282. [PMC free article: PMC8001323] [PubMed: 33805776]
- Xu XL, Singh HP, Wang L, Qi DL, Poulos BK, Abramson DH, Jhanwar SC, Cobrinik D. Rb suppresses human cone-precursor-derived retinoblastoma tumours. Nature. 2014;514:385-8. [PMC free article: PMC4232224] [PubMed: 25252974]
- Zeschnigk M, Böhringer S, Price EA, Onadim Z, Masshöfer L, Lohmann DR. A novel real-time PCR assay for quantitative analysis of methylated alleles (QAMA): analysis of the retinoblastoma locus. Nucleic Acids Res. 2004;32:e125. [PMC free article: PMC519124] [PubMed: 15353561]
Publication Details
Author Information and Affiliations
Institute of Human Genetics
University Hospital Essen
Essen, Germany
Retinoblastoma Program
Hospital for Sick Children
Toronto, Ontario, Canada
Publication History
Initial Posting: July 18, 2000; Last Update: September 21, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Lohmann DR, Gallie BL. Retinoblastoma. 2000 Jul 18 [Updated 2023 Sep 21]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
