Summary
Clinical characteristics.
Individuals with 22q11.2 deletion syndrome (22q11.2DS) can present with a wide range of features that are highly variable, even within families. The major clinical manifestations of 22q11.2DS include congenital heart disease, particularly conotruncal malformations (ventricular septal defect, tetralogy of Fallot, interrupted aortic arch, and truncus arteriosus), palatal abnormalities (velopharyngeal incompetence, submucosal cleft palate, bifid uvula, and cleft palate), immune deficiency, characteristic facial features, and learning difficulties. Hearing loss can be sensorineural and/or conductive. Laryngotracheoesophageal, gastrointestinal, ophthalmologic, central nervous system, skeletal, and genitourinary anomalies also occur. Psychiatric illness and autoimmune disorders are more common in individuals with 22q11.2DS.
Diagnosis/testing.
The diagnosis of 22q11.2DS is established by identification of a heterozygous deletion at chromosome 22q11.2 on chromosomal microarray analysis or other genomic analyses.
Management.
Treatment of manifestations: Cardiac anomalies are treated as recommended by cardiologist; surgical repair for palate anomalies as recommended by otolaryngologist; feeding issues are treated with modification of spoon placement; standard treatment for gastroesophageal reflux and gastrointestinal dysmotility; immune deficiency requires aggressive treatment of infections; rarely, prophylactic antibiotics, IVIG therapy, or thymus tissue implantation are required; irradiated blood products are recommended until normalization of the immune system can be confirmed; treatment of autoimmune disease as per immunologist; calcium supplementation and referral to an endocrinologist and nephrologist because of increased risk of renal calculi if long-term supplementation is required; standard treatment for growth hormone deficiency; standard treatment for ocular anomalies; hearing aids may be helpful for hearing loss; occupational, physical, and speech therapy with introduction of sign language by age one year, educational and behavioral therapy; support and treatment for psychiatric disease as indicated; activity restriction as recommended by an orthopedist for cervical spine anomalies; surgery and treatment as recommended by a nephrologist for renal anomalies; routine dental treatment with consideration of sealants.
Surveillance: Evaluation for nasal speech quality after language emergence; antibody studies to assess seroconversion; reevaluate immune status in childhood before administration of live vaccines; annual complete blood count and differential; serum ionized calcium every three to six months in infancy, every five years through childhood, every one to two years thereafter, preoperatively and postoperatively, and regularly during pregnancy; TSH and free T4 annually; ophthalmologic evaluation between age one and three years or as indicated; audiology evaluation in infancy, at preschool age, and in school age children; developmental assessments annually; annual clinical surveillance for scoliosis; dental examination every six months.
Agents/circumstances to avoid: Infants with lymphocyte abnormalities should not be immunized with live vaccines (e.g., oral polio, MMR). Carbonated drinks and alcohol consumption may exacerbate hypocalcemia. Caffeine intake may contribute to or worsen anxiety.
Genetic counseling.
22q11.2DS is an autosomal dominant contiguous gene deletion syndrome. In 22q11.2DS caused by a 3.0 (2.54)-Mb deletion, the deletion is de novo in more than 90% of individuals and inherited from a heterozygous parent in about 10% of individuals. Sixty percent of individuals with 22q11.2DS caused by a nested 22q11.2 deletion inherited the deletion from an affected parent. Offspring of affected individuals have a 50% chance of inheriting the 22q11.2 deletion. Once the 22q11.2 deletion has been identified in an affected family member, prenatal testing using FISH, MLPA, or array studies for a pregnancy at increased risk and preimplantation genetic testing are possible.
GeneReview Scope
Table
DiGeorge syndrome Velocardiofacial syndrome
Diagnosis
Suggestive Findings
22q11.2 deletion syndrome (22q11.2DS) should be suspected in individuals with the following clinical findings.
Clinical features
- Congenital heart disease (in 64% of individuals), particularly conotruncal defects (e.g., ventricular septal defect, tetralogy of Fallot, interrupted aortic arch, truncus arteriosus)
- Palatal abnormalities (in 67%) including velopharyngeal insufficiency, submucosal cleft palate, bifid uvula, cleft palate, and hypernasal speech, dysphagia
- Laryngotracheoesophageal abnormalities including vascular ring, laryngeal web, laryngotracheomalacia, and subglottic stenosis
- Gastrointestinal anomalies including constipation with or without structural gastrointestinal anomalies (e.g., anteriorly placed/imperforate anus, esophageal atresia, jejunal atresia, intestinal malrotation, Hirschsprung disease), accessory spleens, diaphragmatic hernia, umbilical hernia, and inguinal hernia
- Immune deficiency (in 77%) (e.g., frequent infections, thymic hypoplasia)
- Autoimmune disorders (e.g., juvenile rheumatoid arthritis, Graves disease, vitiligo)
- Ophthalmologic findings including tortuous retinal vessels, ptosis, posterior embryotoxon, sclerocornea, coloboma, cataract, anophthalmia, and strabismus
- Other craniofacial features (e.g., hooded eyelids, ear anomalies, prominent nasal bridge, bulbous nose, micrognathia, asymmetric crying facies, craniosynostosis)
- Hearing loss (sensorineural and/or conductive)
- CNS abnormalities including hypotonia in infancy, microcephaly, polymicrogyria, and seizures (idiopathic or associated with hypocalcemia)
- Developmental delay and/or learning difficulties (in 70%-90%), especially a nonverbal learning disability
- Psychiatric illness including autism spectrum disorder (20% of children), schizophrenia (25% of adults), attention deficit disorder, anxiety, perseveration, and difficulty with social interactions
- Early-onset Parkinson disease
- Skeletal anomalies including occipital-cervical anomaly, scoliosis, rib and vertebral anomalies, clubfoot, and polydactyly
- Genitourinary tract anomalies including renal anomalies (in 16%) (e.g., hydronephrosis, renal agenesis, multicystic/dysplastic kidney), cryptorchidism, and hypospadias
NOTE: See the National Human Genome Research Institute (NHGRI) Atlas of Human Malformation Syndromes (scroll to ATLAS IMAGES) for photographs of individuals with 22q11.2 deletion syndrome from diverse ethnic backgrounds.
Laboratory features
- Hypoparathyroidism and hypocalcemia (in 50%)
- Growth hormone deficiency
- Hypothyroidism
- Cytopenias (hemolytic anemia, neutropenia, thrombocytopenia)
Establishing the Diagnosis
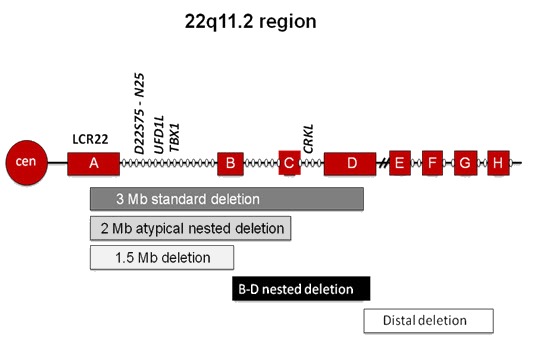
The diagnosis of 22q11.2DS is established in a proband by identification of a heterozygous deletion at chromosome 22q11.2 (see Table 1). The majority of individuals (~85%) with 22q11.2DS have a heterozygous 2.54-Mb deletion, as described based on chromosomal microarray (CMA) designs, at the approximate position of chr22:g.18912231-21465672 in the reference genome (NCBI Build GRCh37/hg19) extending from flanking low copy number repeats (LCRs) A-D and including TBX1 (see Figure 1). Note: Historically, the recurrent deletion has been described as a 3-Mb deletion [Guo et al 2016].
Figure 1.
The majority of affected individuals (85%) have a 2.54-Mb deletion encompassing approximately 40 genes; a subset of individuals have a smaller atypical or "nested" deletion. Adapted from McDonald-McGinn & Zackai [2008]
Note: Approximately 5% of individuals with 22q11.2DS have a heterozygous 1.5-Mb deletion extending from LCRs A-B; about 2% have a deletion extending from LCRs A-C; Approximately 5% have a smaller heterozygous deletion extending from LCRs B-D or C-D.
ISCN nomenclature for this deletion:
- 2.54-Mb del: seq[GRCh37] del(22)(q11.2) chr22:18,912,231-21,465,672
- 1.5-Mb del: seq[GRCh37] del(22)(q11.2) chr22:20,731,986-21,465,672
Note: (1) Since these deletions are recurrent and mediated by segmental duplications, the unique genetic sequence that is deleted is the same in all individuals with each deletion; however, the reported size of the deletion may: (a) may be larger if adjacent segmental duplications are included in the size; and (b) may vary based on the design of the microarray used to detect it (see Molecular Pathogenesis). (2) The phenotype of significantly larger or smaller deletions within this region may be clinically distinct from 22q11.2DS (see Genetically Related Disorders). (3) Although pathogenic variants in a single gene in the 22q11.2 region are not causative of 22q11.2DS, several genes of interest have been identified (see Differential Diagnosis and Molecular Genetics).
Genomic testing methods that determine the copy number of sequences can include chromosomal microarray (CMA) or targeted deletion analysis. Note: The 22q11.2 recurrent deletion cannot be identified by routine analysis of G-banded chromosomes or other conventional cytogenetic banding techniques.
- CMA using oligonucleotide or SNP arrays can detect the recurrent deletion in a proband. The ability to size the deletion depends on the type of microarray used and the density of probes in the 22q11.2 region.Note: (1) Most individuals with a 22q11.2 recurrent deletion are identified by CMA performed in the context of evaluation for developmental delay, intellectual disability, or autism spectrum disorder. (2) Prior to 2004 many CMA platforms did not include coverage for this region and thus may not have detected this deletion. The early arrays used to detect the 22q11.2 deletion were BAC CGH arrays with approximately 25 kb or less for resolution [Mantripragada et al 2004].
- Targeted deletion analysis. FISH analysis, quantitative PCR (qPCR), multiplex ligation-dependent probe amplification (MLPA), or other targeted quantitative methods may be used to test relatives of a proband known to have the 22q11.2 recurrent deletion.Note: (1) Targeted deletion testing is not appropriate for an individual in whom the 22q11.2 recurrent deletion was not detected by CMA designed to target this region. (2) It is possible to size the deletion routinely by use of targeted methods; in particular, MLPA can be used to detect the different deletion sizes by LCR.
Table 1.
Genomic Testing Used in 22q11.2 Deletion Syndrome
Evaluating at-risk relatives. FISH, qPCR, MLPA, or other quantitative methods of targeted deletion analysis can be used to identify the 22q11.2 recurrent deletion in at-risk relatives of the proband. Testing of parental samples is important in determining recurrence risk (see Genetic Counseling).
Clinical Characteristics
Clinical Description
This section summarizes findings based on publications of individuals with 22q11.2 deletion syndrome (22q11.2DS).
Heart. A recent record review of 1,421 individuals with 22q11.2DS revealed congenital heart defects (CHD) in 64% of individuals [Campbell et al 2018]. The most frequent anomalies are conotruncal defects of the outflow tract (see Table 2). Ventricular septal defects were the most common abnormality identified on echocardiography. CHD are the major cause of mortality (~87% of all deaths) in children with 22q11.2DS [McDonald-McGinn et al 2015]. In addition, adults with 22q11.2DS die prematurely, with sudden death and heart failure being the most common causes of death, even in individuals without CHD [Bassett et al 2009]. A subset of affected individuals are found to have dilated aortic root [John et al 2009]. The natural history of aortic root dilatation is unknown.
Table 2.
Cardiac Findings in Individuals with 22q11.2 Deletion Syndrome
Palate. In a review of 1,048 individuals with 22q11.2DS, 67% of individuals had a palatal abnormality (Table 3) [Jackson et al 2019] – a finding consistent with previous studies. The most common abnormality, velopharyngeal incompetence (VPI), may be a structural problem (short palate), a functional problem (hypotonia of the velopharyngeal musculature), or a combination of the two. Submucosal cleft palate and/or a bifid uvula are also fairly prevalent, whereas overt cleft palate and cleft lip/palate are less frequently observed. Often children initially diagnosed with 22q11.2DS because of a cardiac defect are subsequently found to have unrecognized but clinically significant VPI [McDonald-McGinn et al 1997]. It is important to note that the reported incidence of palatal abnormalities varies widely, depending on numerous factors including the reporting technique, the diligence with which the diagnosis is sought, the age at which the individual is evaluated, and the inherent ascertainment bias of any single center. About 26.6% of persons have no palatal involvement.
Table 3.
Palatal Findings with 22q11.2 Deletion Syndrome
Feeding. About 36% of children have significant feeding difficulties, often severe dysphagia requiring nasogastric tube feedings and/or gastrostomy tube placement. Feeding difficulties have been reported in individuals without cardiac defects or palatal anomalies. Further evaluation of such children often reveals a preponderance of nasopharyngeal reflux, prominence of the cricopharyngeal muscle, abnormal cricopharyngeal closure, and/or diverticulum. Thus, the underlying feeding problem in many children appears to be dysmotility in the pharyngoesophageal area, which is derived from the third and fourth pharyngeal pouches. Aspiration should be considered a possible cause for respiratory compromise or recurrent pulmonary infections and reactive airway disease [Eicher et al 2000].
Constipation is a chronic feature in the majority of individuals. In addition, structural anomalies such as imperforate anus, intestinal malrotation, intestinal non-rotation, congenital diaphragmatic hernia, esophageal atresia, tracheoesophageal fistula, Hirschsprung disease, and feeding difficulties secondary to a vascular ring have all been reported and can contribute to significant feeding and swallowing problems and in some instances to constipation [Digilio et al 1999, Kilic et al 2003, Jyonouchi et al 2009, Campbell et al 2018].
Immune function. Immunodeficiency occurs as a result of thymic hypoplasia and subsequent impaired T-cell production. Newborns with 22q11.2DS have significantly fewer cells of thymic lineage. In previous studies, 67% of individuals had impaired T-cell production and 19% had impaired T-cell function [Smith et al 1998, Sullivan et al 1999, Sullivan 2019]. In a study of 1,421 individuals with 22q11.2DS, 50% had abnormal T-cell populations [Campbell et al 2018]. Improvement in T-cell production occurs over time and children with the most significant deficiencies in T-cell production improved most in the first year of life [Sullivan et al 1999]. Individuals with slight decreases in T-cell numbers typically had normal defenses against pathogens [Sullivan 2019].
CD4+ lymphopenia is associated with TBX1 deletions. In a study of 52 infants approximately age one year, CD3 and CD4 counts were significantly lower in infants with a TBX1 deletion (proximal deletions; A-B, A-C, A-D) than in those who did not have a TBX1 deletion (nested and distal deletions; B-D, C-D, D-E, D-F) [Crowley et al 2018].
Abnormalities of humoral immunity were observed in 17% of individuals [Campbell et al 2018]. IgA deficiency was reported in 13% and appears to be particularly common in those with autoimmune problems including juvenile rheumatoid arthritis (JRA) [Sullivan 2019]. Hypogammaglobulinemia present in the first year of life usually resolves and hypergammaglobulinemia may occur after age five. Although the majority of affected individuals have normal antibody function and antibody avidity, some have functional antibody defects. Those with recurrent sinopulmonary infections frequently have immunoglobulin abnormalities, in particular impaired antibody responses to pneumococcal polysaccharide vaccine [Gennery et al 2002, Sullivan 2019].
Palatal dysfunction, gastroesophageal reflux, and aspiration pneumonia can all contribute to recurrent infection, especially in persons with congenital heart disease. Furthermore, dysphagia can lead to poor nutrition, which further impairs cellular immunity. Thus, older children and adults continue to have infections, including 25%-33% with recurrent sinusitis or otitis media and 4%-7% with recurrent lower respiratory infections [Jawad et al 2001]. However, despite these issues, very few school-aged children require active management for their immunodeficiency [Sullivan 2019].
Autoimmune disease. Secondary consequences related to T-cell lymphopenia include an increased risk of atopy and autoimmune disease. JRA occurs in children with 22q11.2DS at a frequency 20 times that in the general population. The age of onset of JRA ranges from 17 months to five years. JRA is often polyarticular and may be difficult to manage. HLA types permissive for the development of JRA are observed [Sullivan et al 1997]. Other autoimmune disorders associated with 22q11.2DS include: idiopathic thrombocytopenia purpura (ITP), hyperthyroidism (Graves disease), hypothyroidism, vitiligo, hemolytic anemia, autoimmune neutropenia, aplastic anemia, and celiac disease. ITP is seen 200 times more frequently in individuals with 22q11.2DS than in the general population [Jawad et al 2001, Kawame et al 2001, Sullivan 2019].
Parathyroid function. Hypoparathyroidism and subsequent hypocalcemia is present in 17%-60% of persons with 22q11.2DS and is typically most serious in the neonatal period. Calcium homeostasis often normalizes with age, although recurrence of hypocalcemia in later childhood and adulthood has been reported during illness, puberty, and pregnancy. Cheung et al [2014] reported that 80% of adults with 22q11.2DS experienced hypocalcemia sometime during their lifetime, and that hypoparathyroidism, hypothyroidism, and hypomagnesemia may contribute to hypocalcemia.
Growth. Most adults with 22q11.2DS are of normal stature; however, in 95 children between age one and 15 years, 41% were below the fifth percentile in height. Of these, four were significantly below the fifth percentile; all four individuals had low concentrations of IGF1 and IGFBP3. Three had evidence of growth hormone deficiency; three had a small pituitary gland on MRI; and two responded to human growth hormone therapy [Weinzimer et al 1998]. Growth charts specific to 22q11.2DS have been developed [Habel et al 2012]. Obesity has been reported in up to 35% of adults with 22q11.2DS [Bassett et al 2005].
Eyes. A prospective evaluation for ocular abnormalities in 90 individuals revealed hooding lids (20%), ptosis (4%), and distichiasis (abnormal growth of lashes from the orifices of the meibomian glands) (2%). Other findings included posterior embryotoxon (49%), prominent corneal nerves (3%), prominent iris crypts (2%), tortuous retinal vessels (34%), and tilted optic nerves (1%). Strabismus was observed in 18% and amblyopia in 4%. A small number of persons have cataracts and colobomas [Forbes et al 2007]. While posterior embryotoxon was observed in 12%-32% of controls, the incidence in individuals with 22q11.2DS was almost as high as that seen in Alagille syndrome (89%) [Krantz et al 1997]. The incidence of astigmatism, myopia, and hyperopia was comparable to that in the general population. Anophthalmia has been observed in a very small subset of individuals [Author, unpublished data].
Other craniofacial features. In addition to palate and ocular anomalies craniofacial findings can include ear anomalies, nasal anomalies, asymmetric crying facies, micrognathia, and craniosynostosis [McDonald-McGinn et al 2015]. Ear abnormalities include overfolded or squared off helices; cupped, microtic, and protuberant ears; preauricular pits or tags; and narrow external auditory meati. Sensorineural and conductive hearing loss have both been reported. A prominent nasal bridge, bulbous nose, hypoplastic alae nasi, and nasal dimple / bifid nasal tip are common [Campbell et al 2018]. Stridor resulting from vascular ring, laryngomalacia, and laryngeal web, laryngeal atresia, and subglottic stenosis can occur. Facial features are variable and may not be present especially in persons of African American descent [Kruszka et al 2017].
Central nervous system. The majority of individuals with 22q11.2DS have a history of hypotonia in infancy [Swillen & McDonald-McGinn 2015]. Studies report asymmetric crying facies in 8%-14% of individuals [McDonald-McGinn et al 1997, Campbell et al 2018]. Microcephaly has been reported in 24%-50% of affected individuals [McDonald-McGinn & Sullivan 2011, Campbell et al 2018]. Seizures are most often associated with hypocalcemia. However, approximately 15% of persons with 22q11.2DS had unprovoked seizures [Fung et al 2015].
A study of 24 individuals with 22q11.2DS and MRI/ MRA showed more than half (13/24) had significant radiographic findings, including persistent cavum septi pellucidi and/or cavum vergae (8/24), polymicrogyria or cortical dysplasia (4/24), and hypoplastic cerebellum (1/24) [Bohm et al 2017]. Functional MRI scans revealed significantly reduced posterior brain volumes relative to age- and sex-matched controls with more significant white matter loss in the left occipital and left parietal regions than in the frontal lobes [Bearden et al 2004, Bish et al 2004, Kates et al 2004]. A large multi-site study using diffusion tensor imaging indicated widespread reductions in mean, axial, and radial diffusivities in individuals with 22q11.2DS especially in major cortico-cortical connections [Villalón-Reina et al 2020].
Psychosocial development and cognitive function. In general, young children with 22q11.2DS have delays in motor milestones with a mean age at walking of 18 months, and delay in emergence of language (many are nonverbal at age 2-3 years).
In 55 toddlers assessed with Bayley Scales, mental development was average in 22%, mildly delayed in 20%, and significantly delayed in 58%. The mean mental developmental index was 67±15, which falls in the significantly delayed range, and the mean psychomotor developmental index was 61±13. Speech and language delays were present in all children assessed. In the same study, in a group of 24 preschoolers assessed using the WPPSI-R, the full-scale IQ was 78±12, the mean performance IQ was 78±12, and the mean verbal IQ was 81±13. In total language, 20% were average, 46% were mildly delayed, and 34% were significantly delayed with receptive language scores higher than expressive [Gerdes et al 2001].
In a group of 80 school-aged children assessed with the age-appropriate Weschler IQ test, the mean IQ score was 76.8±13.0; 39% attained full-scale IQ scores in the average range, 31% in the low-average range, and 31% in the borderline range [Niarchou et al 2014].
Older individuals with 22q11.2DS generally have an atypical neuropsychologic profile across multiple domains, the most striking aspect of which is a significantly higher verbal IQ score than performance IQ score. Moss et al [1995] observed a mean split between the verbal IQ and performance IQ in 66% of 80 school-age children consistent with a nonverbal learning disability that is rare in the general population [Swillen et al 2018]. Because the full-scale IQ score alone does not accurately represent the abilities of many individuals with 22q11.2DS, verbal and performance IQ scores need to be considered separately. In addition, affected individuals exhibit relative strengths in the areas of rote verbal learning and memory, reading decoding, and spelling. Deficits are found in the areas of nonverbal processing, visual-spatial skills, complex verbal memory, attention, working memory, visual-spatial memory, and mathematics. This evidence of stronger verbal-than-visual memory skills and stronger reading-than-math skills also supports the presence of a nonverbal learning disorder that requires specific cognitive remediation, behavior management, and parental counseling.
Psychiatric illness. Behavior and temperament observed in some individuals with 22q11.2DS include disinhibition and impulsiveness on the one hand and shyness and withdrawal on the other. Attention deficit, anxiety, perseveration, and difficulty with social interactions are also common. Autism / autism-spectrum disorders are reported in approximately 20% of individuals [Swillen & McDonald-McGinn 2015].
It has been suggested that 60% of adults have a psychiatric disorder. Most notably, schizophrenia is identified in approximately 25% of individuals; however, anxiety and depressive disorders are also quite common [Bassett et al 2011]. Behavioral differences may begin at a young age; screening children with 22q11.2DS for psychiatric issues before age ten years may provide an opportunity for early intervention [Swillen & McDonald-McGinn 2015].
Early-onset Parkinson disease. 22q11.2DS is associated with an increased risk of early-onset Parkinson disease with a reported prevalence of 5.9% [Fung et al 2015]. To date, there are few studies of other neurodegenerative disorders in 22q11.2DS.
Musculoskeletal system. A review of the musculoskeletal manifestations in 22q11.2DS revealed strong evidence that 90.5%-100% of individuals with occiput and cervical imaging (10 studies including 408 individuals) had at least one occipital-cervical anomaly [Homans et al 2018]. Common features included flattening of the skull, dysmorphic shape or an open arch at the first cervical vertebra, and dysmorphic dens with upswept lamina and posterior elements at the second vertebra. A frequently reported anomaly on flexion-extension radiographs is increased segmental motion (56%). Scoliosis was described in 14 studies (2,264 individuals), with a prevalence of 0.6%-60%. Rib anomalies were noted in two studies with a prevalence of 2%-19% and vertebral differences were observed in 1.1%-11%.
The most common extremity manifestation is pes equinovarus (clubfoot) which was described in 15 studies (2,115 individuals), with a prevalence of 1.1%-13.3%. Patellar dislocation was observed in three studies with a prevalence of 10%-20%, though this evidence was weaker. Other anomalies reported included polydactyly, shoulder deformities, and overfolded toes.
Genitourinary. Renal anomalies were identified in 16% of individuals with 22q11.2DS; the most common anomalies included unilateral hydronephrosis, renal agenesis, and multicystic dysplastic kidney [Campbell et al 2018]. Although hydronephrosis was the most common upper tract finding, the majority (63%) had isolated upper tract dilatation. Boys were significantly more likely to be diagnosed with a genitourinary abnormality than girls (8% vs 0.5%). Among males, 4% had cryptorchidism and 4% had hypospadias. Additional anomalies in females included vaginal agenesis in two individuals and uterine agenesis in one individual. Other reported abnormalities included umbilical hernia, inguinal hernia, chordee, phimosis, and undescended testes.
Other. Other findings observed in individuals with 22q11.2DS:
- Abnormal lung lobation [McDonald-McGinn et al 1995]
- Dental carries
- Malignancies including hepatoblastoma, renal cell carcinoma, thyroid carcinoma, melanoma, leukemia, Wilms tumor, and neuroblastoma [Campbell et al 2018, Lambert et al 2018]; overall prevalence approximately 6%
- Autosomal recessive disorders (reported in individuals with 22q11.2DS and a pathogenic variant of the second allele) including Bernard-Soulier syndrome (caused by a pathogenic variant in GP1BB) and CEDNIK syndrome (caused by a pathogenic variant in SNAP29) [Campbell et al 2018].
Genotype-Phenotype Correlations
A correlation between CD4+ lymphopenia and deletion breakpoints has been reported. CD3 and CD4 counts were significantly lower in individuals with 22q11.2 proximal deletions (A-B, A-C, A-D) compared to those with nested and distal deletions (B-D, C-D, D-E, D-F) [Crowley et al 2018].
Penetrance
Penetrance is complete in the majority of individuals with 22q11.2DS; variability is marked. Nested deletions are often familial and have reduced penetrance and/or a milder expression.
Nomenclature
It is now recognized that 22q11.2DS encompasses the phenotypes previously described as DiGeorge syndrome (DGS), velocardiofacial syndrome (VCFS), conotruncal anomaly face syndrome (CTAF), some cases of autosomal dominant Opitz G/BBB syndrome, and Cayler cardiofacial syndrome (asymmetric crying facies) [McDonald-McGinn et al 2015]. The clinical descriptions of these entities resulted from an ascertainment bias.
The term DiGeorge syndrome is now reserved for individuals who have clinical features of 22q11.2DS but do not have an identified 22q11.2 deletion.
Autosomal dominant Opitz G/BBB syndrome may also be referred to as hypertelorism with esophageal abnormality and hypospadias.
Prevalence
22q11.2DS is the most frequent chromosome microdeletion syndrome. In a population-based study in Sweden, the mean annual incidence was 14.1:100,000 live births [Oskarsdóttir et al 2004, Oskarsdóttir et al 2005a, Oskarsdóttir et al 2005b]. A US population-based study found an overall prevalence of about 1:6,000 in whites, blacks, and Asians, and 1:3,800 in Hispanics [Botto et al 2003].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with deletion of 22q11.2.
A small percentage (<1%) of individuals with clinical findings of 22q11.2DS have chromosome rearrangements involving 22q11.2, such as a translocation between chromosome 22 and another autosome.
Differential Diagnosis
All of the clinical findings associated with 22q11.2 deletion syndrome (22q11.2DS) can also occur as an isolated anomaly in an otherwise healthy individual. Genetic disorders and teratogenic exposures that may cause a clinical phenotype similar to 22q11.2DS are discussed in this section.
Single-Gene Disorders
Table 4.
Genes of Interest in the Differential Diagnosis of 22q11.2 Deletion Syndrome
Chromosome Disorders
Deletion 10p13-p14. Features overlapping with 22q11.2DS can include cardiac defects, immune deficiency, hypoparathyroidism, cleft palate, developmental delay, microcephaly, and cryptorchidism [Lichtner et al 2000].
Deletion 11q23-ter (Jacobsen syndrome) (OMIM 147791). Features overlapping with 22q11.2DS can include microcephaly, micrognathia, low set ears, ocular manifestations, cardiac defects, hypospadias, cryptorchidism, and immune deficiency.
Other
Disorders of unknown genetic etiology
- VACTERL association (when congenital heart disease, vertebral, renal, and limb anomalies are present). VATER association is a diagnosis of exclusion without an established etiology to date (OMIM 192350).
- Oculoauriculovertebral (Goldenhar) syndrome (OAVS) (when ear anomalies, vertebral defects, heart disease, renal anomalies are present) (OMIM 141400)
Teratogenic exposures. A phenotype similar to 22q11.2DS can be associated with maternal diabetes and maternal retinoic acid exposure [Digilio et al 1995, Coberly et al 1996].
Management
Evaluations Following Initial Diagnosis
Clinical practice guidelines for the evaluation and treatment of individuals with 22q11.2 deletion syndrome (22q11.2DS) have been published. See Bassett et al [2011] (full text) and Fung et al [2015].
To establish the extent of disease and needs of an individual diagnosed with 22q11.2DS, the evaluations summarized in Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 5.
Recommended Evaluations Following Initial Diagnosis in Individuals with 22q11.2 Deletion Syndrome
Treatment of Manifestations
Table 6.
Treatment of Manifestations in Individuals with 22q11.2 Deletion Syndrome
Surveillance
Table 7.
Recommended Surveillance for Individuals with 22q11.2 Deletion Syndrome
Agents/Circumstances to Avoid
Infants with lymphocyte abnormalities should not be immunized with live vaccines (e.g., oral polio, MMR).
Carbonated drinks and alcohol consumption may exacerbate hypocalcemia.
Caffeine intake may contribute to or worsen anxiety.
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic sibs and parents of an affected individual in order to identify as early as possible those family members who would benefit from cardiac and immunologic evaluation and evaluations and surveillance for other complications of 22q11.2DS.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Pregnant women must be monitored medically, taking into account any preexisting conditions including congenital heart disease, scoliosis, and reactive airway disease. Additional surveillance should include calcium, thyroid, and platelet levels. In addition, individuals with changes in mental status/behavior should be referred for immediate evaluation by a mental health care provider.
A fetus at high risk of having 22q11.2DS should undergo a level II ultrasound with fetal echocardiogram to evaluate for the following anomalies: congenital heart disease; airway, palate, swallowing, and gastrointestinal differences possibly leading to polyhydramnios (congenital diaphragmatic hernia, tracheoesophageal fistula, subglottic stenosis, vascular ring, laryngeal web, cleft palate, and cleft and lip/palate); renal anomalies; skeletal differences such as club foot and craniosynostosis; and umbilical and inguinal hernia.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
22q11.2 deletion syndrome (22q11.2DS) is a contiguous gene deletion syndrome inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- In 22q11.2DS caused by a 3.0 (2.54)-Mb deletion, the deletion is de novo in more than 90% of individuals and inherited from a heterozygous parent in about 10% of individuals [Bassett et al 2011, McDonald-McGinn et al 2015].
- The proportion of individuals with a nested deletion (i.e., a recurrent, atypical shorter deleted segment [LCR22B-D] nested within the large typically deleted region) inherited from an affected parent is higher (60%).
- Genomic testing capable of identifying the deletion identified in the proband is recommended for the parents of a proband in order to reliably determine recurrence risk.
- If the 22q11.2 deletion cannot be detected in the leukocyte DNA of either parent, possible explanations include a de novo deletion in the proband or germline mosaicism in a parent. Parental germline mosaicism has been reported [McDonald-McGinn et al 2015].
- The family history of some individuals diagnosed with 22q11.2DS may appear to be negative because of failure to recognize the disorder in family members because of clinical variability and/or reduced penetrance. Therefore, an apparently negative family history cannot be confirmed unless the parents have been tested for the 22q11.2 deletion identified in the proband.
- Note: If the parent is the individual in whom the pathogenic variant first occurred, the parent may have somatic mosaicism for the variant and may be mildly/minimally affected. Apparently asymptomatic adults with somatic mosaicism have been identified [McDonald-McGinn et al 2015].
Sibs of a proband. The risk to the sibs of a proband depends on the clinical/genetic status of the proband's parents:
- If a parent is affected and/or is known to have a 22q11.2 deletion, the risk to the sibs is 50%. The phenotype in a sib who inherits the deletion cannot be predicted because 22q11.2DS is associated with significant intrafamilial clinical variability.
- If the 22q11.2 deletion identified in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental germline mosaicism [McDonald-McGinn et al 2015].
- If the parents have not been tested for the 22q11.2 deletion but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for 22q11.2DS because of the possibility of reduced penetrance in a heterozygous parent or parental germline mosaicism.
- In the (unlikely) event that a parent has a balanced structural chromosome rearrangement involving the 22q11.2 region, the risk to sibs is increased. The estimated risk depends on the specific chromosome rearrangement.
Offspring of a proband. Each child of an individual with 22q11.2DS has a 50% chance of inheriting the 22q11.2 deletion.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the deletion, the parent's family members may be at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
Prenatal Testing and Preimplantation Genetic Testing
High-risk pregnancies
- Molecular genetic testing. Once the 22q11.2 deletion has been identified in an affected family member, prenatal testing using FISH, MLPA, or array studies for a pregnancy at increased risk and preimplantation genetic testing are possible.
- Ultrasound evaluation. Pregnancies at increased risk may be evaluated between 18 and 22 weeks' gestation by high-resolution ultrasound examination for palatal and other associated anomalies and by echocardiography for cardiac anomalies. Note: Gestational age is expressed as menstrual weeks calculated either from the first day of the last normal menstrual period or by ultrasound measurements.
Low-risk pregnancies. In some pregnancies not known by family history to be at increased risk for 22q11.2DS, findings of congenital heart disease and/or cleft palate or cleft lip/palate detected by routine ultrasound examination may suggest the diagnosis, in particular in those individuals with conotruncal cardiac anomalies such as interrupted aortic arch, truncus arteriosus, tetralogy of Fallot, and ventricular septal defect. Additional structural differences that can be associated with 22q11.2DS and may be identified prenatally include: congenital diaphragmatic hernia, umbilical or inguinal hernia, tracheoesophageal fistula / esophageal atresia / laryngeal atresia, polydactyly, craniosynostosis, polymicrogyria, and renal anomalies. In addition, polyhydramnios is frequently present. Chromosome preparations obtained from fetal cells can be analyzed using array studies, MLPA, or FISH. Establishing the diagnosis of 22q11.2DS even late in gestation can be useful for perinatal management.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- International 22q11.2 Deletion Syndrome Foundation, Inc.P.O. Box 2269Cinnaminson NJ 08077Phone: 877-739-1849 (toll-free)Email: info@22q.org
- Max Appeal15 Meridian AvenueStourbridge West Midlands DY8 1049United KingdomPhone: 0800 389 1049 toll freeEmail: info@maxappeal.org.uk
- Medical Home Portal
- National Library of Medicine Genetics Home Reference
- NCBI Genes and Disease
- Chromosome 22 CentralPhone: 919-762-7979Email: usinfo@c22c.org; c22central@gmail.com
- Chromosome Disorder Outreach Inc.Phone: 561-395-4252Email: info@chromodisorder.org
- European Society for Immunodeficiencies (ESID) RegistryEmail: esid-registry@uniklinik-freiburg.de
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
22q11.2 Deletion Syndrome: Genes and Databases
Table B.
OMIM Entries for 22q11.2 Deletion Syndrome (View All in OMIM)
Molecular Pathogenesis
The 3.0-Mb deletion at chromosome 22q11.2 (referred to in CMA nomenclature as a 2.54-Mb deletion with coordinates ~18912231-21465672, genome build UCSC GRCh37) results in the deletion of several genes of interest (see Genes of interest in this region).
More than 85% of individuals with a 22q11.2 deletion have deletions in the same ~3.0-Mb (2.54-Mb by CMA) region; the remainder have either variant deletion endpoints or recurrent, atypical shorter deleted segments nested within the large typically deleted region (TDR) (see Figure 1) [Levy et al 1995, Kurahashi et al 1996, O'Donnell et al 1997, McQuade et al 1999]:
- 3.0 (2.54)-Mb del: ISCN-37446
- 2-Mb atypical del: ISCN: NA
- 1.5-Mb del: ISCN-37516
- Distal deletion: ISCN-46292
A small 20-kb deletion within the TDR was reported in an individual with a classic VCFS/DGS phenotype [Yamagishi et al 1999]. This smaller deletion disrupts UFD1L and CDC45L. In several additional affected individuals, the deletions do not overlap the typically deleted region in that they begin distal to it and extend toward the telomere. The location of duplicated sequence blocks in the vicinity of the 22q11.2 deletion endpoints strongly implicates them in the events leading to the typical and atypical deletions.
A small number of individuals have the deletion as the result of unbalanced translocations that delete the 22pter → q11 region. (For more information, see Table A, Locus Specific.)
Genes of interest in this region
- TBX1. Loss of TBX1 is associated with congenital heart defects (see Differential Diagnosis, Single-Gene Disorders). In addition, TBX1 has been associated with cardiac, pharyngeal, brain microvasculature, and cognitive and behavioral deficits [McDonald-McGinn et al 2015].
- DGCR8 may have an epigenetic role and modify expression of genes contributing to the neuropsychiatric and other phenotypes associated with 22q11.2DS.
- CRKL has been associated with cardiac anomalies and appears to modulate natural killer cell function.
- SNAP29 pathogenic variants have been associated with CEDNIK syndrome (OMIM 609528), a recessive disorder.
- PRODH pathogenic variants have been associated with hyperprolinemia type I (OMIM 239500), a recessive disorder.
Chapter Notes
Revision History
- 9 May 2024 (ma) Revision: thymus tissue implantation
- 27 February 2020 (sw) Comprehensive update posted live
- 28 February 2013 (me) Comprehensive update posted live
- 16 December 2005 (me) Comprehensive update posted live
- 23 July 2003 (me) Comprehensive update posted live
- 23 September 1999 (me) Review posted live
- 7 August 1998 (dmm) Original submission
References
Literature Cited
- Bassett AS, Chow EW, Husted J, Hodgkinson KA, Oechslin E, Harris L, Silversides C. Premature death in adults with 22q11.2 deletion syndrome. J Med Genet. 2009;46:324-30. [PMC free article: PMC3188306] [PubMed: 19246480]
- Bassett AS, Chow EW, Husted J, Weksberg R, Caluseriu O, Webb GD, Gatzoulis MA. Clinical features of 78 adults with 22q11 deletion syndrome. Am J Med Genet A. 2005;138:307-13. [PMC free article: PMC3127862] [PubMed: 16208694]
- Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, Marino B, Oskarsdottir S, Philip N, Sullivan K, Swillen A, Vorstman J; International 22q11.2 Deletion Syndrome Consortium. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr. 2011;159:332-9.e1. [PMC free article: PMC3197829] [PubMed: 21570089]
- Bearden CE, van Erp TG, Monterosso JR, Simon TJ, Glahn DC, Saleh PA, Hill NM, McDonald-McGinn DM, Zackai E, Emanuel BS, Cannon TD. Regional brain abnormalities in 22q11.2 deletion syndrome: association with cognitive abilities and behavioral symptoms. Neurocase. 2004;10:198-206. [PubMed: 15788257]
- Bish JP, Nguyen V, Ding L, Ferrante S, Simon TJ. Thalamic reductions in children with chromosome 22q11.2 deletion syndrome. Neuroreport. 2004;15:1413-5. [PubMed: 15194864]
- Bohm LA, Zhou TC, Mingo TJ, Dugan SL, Patterson RJ, Sidman JD, Roby BB. Neuroradiographic findings in 22q11.2 deletion syndrome. Am J Med Genet A. 2017;173:2158-65. [PubMed: 28577347]
- Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, Merritt RK, O'Leary LA, Wong LY, Elixson EM, Mahle WT, Campbell RM. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112:101-7. [PubMed: 12837874]
- Campbell IM, Sheppard SE, Crowley TB, McGinn DE, Bailey A, McGinn MJ, Unolt M, Homans JF, Chen EY, Salmons HI, Gaynor JW, Goldmuntz E, Jackson OA, Katz LE, Mascarenhas MR, Deeney VFX, Castelein RM, Zur KB, Elden L, Kallish S, Kolon TF, Hopkins SE, Chadehumbe MA, Lambert MP, Forbes BJ, Moldenhauer JS, Schindewolf EM, Solot CB, Moss EM, Gur RE, Sullivan KE, Emanuel BS, Zackai EH, McDonald-McGinn DM. What is new with 22q? An update from the 22q and You Center at the Children's Hospital of Philadelphia. Am J Med Genet A. 2018;176:2058-69. [PMC free article: PMC6501214] [PubMed: 30380191]
- Cheung EN, George SR, Costain GA, Andrade DM, Chow EW, Silversides CK, Bassett AS. Prevalence of hypocalcaemia and its associated features in 22q11·2 deletion syndrome. Clin Endocrinol (Oxf). 2014;81:190-6. [PMC free article: PMC4231257] [PubMed: 24735350]
- Coberly S, Lammer E, Alashari M. Retinoic acid embryopathy: case report and review of literature. Pediatr Pathol Lab Med. 1996;16:823–36. [PubMed: 9025880]
- Crowley B, Ruffner M, McDonald-McGinn DM, Sullivan KE. Variable immune deficiency related to deletion size in chromosome 22q11.2 deletion syndrome. Am J Med Genet A. 2018;176:2082-6. [PMC free article: PMC6470357] [PubMed: 29341423]
- Digilio MC, Marino B, Bagolan P, Giannotti A, Dallapiccola B. Microdeletion 22q11 and oesophageal atresia. J Med Genet. 1999;36:137-9. [PMC free article: PMC1734297] [PubMed: 10051013]
- Digilio MC, Marino B, Formigari R, Giannotti A. Maternal diabetes causing DiGeorge anomaly and renal agenesis. Am J Med Genet. 1995;55:513–4. [PubMed: 7762600]
- Eicher PS, McDonald-McGinn DM, Fox CA, Driscoll DA, Emanuel BS, Zackai EH. Dysphagia in children with a 22q11.2 deletion: unusual pattern found on modified barium swallow. J Pediatr. 2000;137:158-64. [PubMed: 10931405]
- Forbes BJ, Binenbaum G, Edmond JC, DeLarato N, McDonald-McGinn DM, Zackai EH. Ocular findings in the chromosome 22q11.2 deletion syndrome. J AAPOS. 2007;11:179-82. [PubMed: 17140829]
- Fung WL, Butcher NJ, Costain G, Andrade DM, Boot E, Chow EW, Chung B, Cytrynbaum C, Faghfoury H, Fishman L, García-Miñaúr S, George S, Lang AE, Repetto G, Shugar A, Silversides C, Swillen A, van Amelsvoort T, McDonald-McGinn DM, Bassett AS. Practical guidelines for managing adults with 22q11.2 deletion syndrome. Genet Med. 2015;17:599-609. [PMC free article: PMC4526275] [PubMed: 25569435]
- Gennery AR, Barge D, O'Sullivan JJ, Flood TJ, Abinun M, Cant AJ. Antibody deficiency and autoimmunity in 22q11.2 deletion syndrome. Arch Dis Child. 2002;86:422-5. [PMC free article: PMC1763000] [PubMed: 12023174]
- Gerdes M, Solot C, Wang PP, McDonald-McGinn DM, Zackai EH. Taking advantage of early diagnosis: preschool children with the 22q11.2 deletion. Genet Med. 2001;3:40-4. [PubMed: 11339376]
- Gong W, Gottlieb S, Collins J, Blescia A, Dietz H, Goldmuntz E, McDonald-McGinn DM, Zackai EH, Emanuel BS, Driscoll DA, Budarf ML. Mutation analysis of TBX1 in non-deleted patients with features of DGS/VCFS or isolated cardiovascular defects. J Med Genet. 2001;38:E45. [PMC free article: PMC1734783] [PubMed: 11748311]
- Guo X, Delio M, Haque N, Castellanos R, Hestand MS, Vermeesch JR, Morrow BE, Zheng D. Variant discovery and breakpoint region prediction for studying the human 22q11.2 deletion using BAC clone and whole genome sequencing analysis. Hum Mol Genet. 2016;25:3754-67. [PMC free article: PMC5216616] [PubMed: 27436579]
- Gupton SE, McCarthy EA, Markert ML. Care of children with DiGeorge before and after cultured thymus tissue implantation. J Clin Immunol. 2021;41:896-905. [PMC free article: PMC8249267] [PubMed: 34003433]
- Habel A, McGinn MJ 2nd, Zackai EH, Unanue N, McDonald-McGinn DM. Syndrome-specific growth charts for 22q11.2 deletion syndrome in Caucasian children. Am J Med Genet A. 2012;158A:2665-71. [PubMed: 22711268]
- Homans JF, Tromp IN, Colo D, Schlösser TPC, Kruyt MC, Deeney VFX, Crowley TB, McDonald-McGinn DM, Castelein RM. Orthopaedic manifestations within the 22q11.2 deletion syndrome: a systematic review. Am J Med Genet A. 2018;176:2104-20. [PubMed: 29159873]
- Jackson O, Crowley TB, Sharkus R, Smith R, Jeong S, Solot C, McDonald-Mcginn D. Palatal evaluation and treatment in 22q11.2 deletion syndrome. Am J Med Genet A. 2019;179:1184-95. [PubMed: 31038278]
- Jawad AF, McDonald-Mcginn DM, Zackai E, Sullivan KE. Immunologic features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). J Pediatr. 2001;139:715-23. [PubMed: 11713452]
- John AS, McDonald-McGinn DM, Zackai EH, Goldmuntz E. Aortic root dilation in patients with 22q11.2 deletion syndrome. Am J Med Genet A. 2009;149A:939-42. [PMC free article: PMC4080309] [PubMed: 19353635]
- Jyonouchi S, McDonald-McGinn DM, Bale S, Zackai EH, Sullivan KE. CHARGE (coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, ear anomalies/deafness) syndrome and chromosome 22q11.2 deletion syndrome: a comparison of immunologic and nonimmunologic phenotypic features. Pediatrics. 2009;123:e871-7. [PMC free article: PMC4098848] [PubMed: 19403480]
- Kates WR, Burnette CP, Bessette BA, Folley BS, Strunge L, Jabs EW, Pearlson GD. Frontal and caudate alterations in velocardiofacial syndrome (deletion at chromosome 22q11.2). J Child Neurol. 2004;19:337-42. [PubMed: 15224707]
- Kawame H, Adachi M, Tachibana K, Kurosawa K, Ito F, Gleason MM, Weinzimer S, Levitt-Katz L, Sullivan K, McDonald-McGinn DM. Graves' disease in patients with 22q11.2 deletion. J Pediatr. 2001;139:892-5. [PubMed: 11743521]
- Kilic SS, Gurpinar A, Yakut T, Egeli U, Dogruyol H. Esophageal atresia and tracheo-esophageal fistula in a patient with Digeorge syndrome. J Pediatr Surg. 2003;38:E21-3. [PubMed: 12891520]
- Krantz ID, Piccoli DA, Spinner NB. Alagille syndrome. J Med Genet. 1997;34:152-7. [PMC free article: PMC1050871] [PubMed: 9039994]
- Kruszka P, Addissie YA, McGinn DE, Porras AR, Biggs E, Share M, Crowley TB, Chung BH, Mok GT, Mak CC, Muthukumarasamy P, Thong MK, Sirisena ND, Dissanayake VH, Paththinige CS, Prabodha LB, Mishra R, Shotelersuk V, Ekure EN, Sokunbi OJ, Kalu N, Ferreira CR, Duncan JM, Patil SJ, Jones KL, Kaplan JD, Abdul-Rahman OA, Uwineza A, Mutesa L, Moresco A, Obregon MG, Richieri-Costa A, Gil-da-Silva-Lopes VL, Adeyemo AA, Summar M, Zackai EH, McDonald-McGinn DM, Linguraru MG, Muenke M. 22q11.2 deletion syndrome in diverse populations. Am J Med Genet A. 2017;173:879-88. [PMC free article: PMC5363275] [PubMed: 28328118]
- Kurahashi H, Nakayama T, Osugi Y, Tsuda E, Masuno M, Imaizumi K, Kamiya T, Sano T, Okada S, Nishisho I. Deletion mapping of 22q11 in CATCH22 syndrome: identification of a second critical region. Am J Hum Genet. 1996;58:1377–81. [PMC free article: PMC1915078] [PubMed: 8651317]
- Lambert MP, Arulselvan A, Schott A, Markham SJ, Crowley TB, Zackai EH, McDonald-McGinn DM. The 22q11.2 deletion syndrome: cancer predisposition, platelet abnormalities and cytopenias. Am J Med Genet A. 2018;176:2121-7. [PubMed: 28940864]
- Levy A, Demczuk S, Aurias A, Depetris D, Mattei MG, Philip N. Interstitial 22q11 microdeletion excluding the ADU breakpoint in a patient with DiGeorge syndrome. Hum Mol Genet. 1995;4:2417–9. [PubMed: 8634722]
- Lichtner P, Konig R, Hasegawa T, Van Esch H, Meitinger T, Schuffenhauer S. An HDR (hypoparathyroidism, deafness, renal dysplasia) syndrome locus maps distal to the DiGeorge syndrome region on 10p13/14. J Med Genet. 2000;37:33-7. [PMC free article: PMC1734454] [PubMed: 10633131]
- Mantripragada KK, Tapia-Páez I, Blennow E, Nillson P, Wedell A, Dumanski JP. DNA copy-number analysis of the 22q11 deletion-syndrome region using array-CGH with genomic and PCR-based targets. Int J Mol Med. 2004;13:273-9. [PubMed: 14719134]
- McDonald-McGinn DM, Driscoll DA, Bason L, Christensen K, Lynch D, Sullivan K, Canning D, Zavod W, Quinn N, Rome J. Autosomal dominant "Opitz" GBBB syndrome due to a 22q11.2 deletion. Am J Med Genet. 1995;59:103-13. [PubMed: 8849001]
- McDonald-McGinn DM, LaRossa D, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, Moss E, Wang P, Solot C, Schultz P, Lynch D, Bingham P, Keenan G, Weinzimer S, Ming JE, Driscoll D, Clark BJ 3rd, Markowitz R, Cohen A, Moshang T, Pasquariello P, Randall P, Emanuel BS, Zackai EH. The 22q11.2 deletion: screening, diagnostic workup, and outcome of results; report on 181 patients. Genet Test. 1997;1:99-108. [PubMed: 10464633]
- McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vortsman JA, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, Scambler PJ, Bassett AS. 22q11.2 deletion syndrome. Nat Rev Dis Primers. 2015;1:15071. [PMC free article: PMC4900471] [PubMed: 27189754]
- McDonald-McGinn DM, Sullivan KE. Chromosome 22q11. 2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine. 2011;90:1–18. [PubMed: 21200182]
- McDonald-McGinn DM, Zackai EH. Genetic counseling for the 22q11.2 deletion. Dev Disabil Res Rev. 2008;14:69-74. [PubMed: 18636638]
- McQuade L, Christodoulou J, Budarf M, Sachdev R, Wilson M, Emanuel B, Colley A. Patient with a 22q11.2 deletion with no overlap of the minimal DiGeorge syndrome critical region (MDGCR). Am J Med Genet. 1999;86:27–33. [PubMed: 10440825]
- Moss E, Wang PP, McDonald-McGinn DM, et al. Characteristic cognitive profile in patients with a 22q11 deletion: verbal IQ exceeds nonverbal IQ. Am J Hum Genet. 1995;57:A42.
- Niarchou M, Zammit S, van Goozen SH, Thapar A, Tierling HM, Owen MJ, van den Bree MB. Psychopathology and cognition in children with 22q11.2 deletion syndrome. Br J Psychiatry. 2014;204:46–54. [PMC free article: PMC3877833] [PubMed: 24115343]
- O'Donnell H, McKeown C, Gould C, Morrow B, Scambler P. Detection of an atypical 22q11 deletion that has no overlap with the DiGeorge syndrome critical region. Am J Hum Genet. 1997;60:1544. [PMC free article: PMC1716117] [PubMed: 9199579]
- Oskarsdóttir S, Belfrage M, Sandstedt E, Viggedal G, Uvebrant P. Disabilities and cognition in children and adolescents with 22q11 deletion syndrome. Dev Med Child Neurol. 2005a;47:177–84. [PubMed: 15739722]
- Oskarsdóttir S, Persson C, Eriksson BO, Fasth A. Presenting phenotype in 100 children with the 22q11 deletion syndrome. Eur J Pediatr. 2005b;164:146–53. [PubMed: 15565286]
- Oskarsdóttir S, Vujic M, Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Arch Dis Child. 2004;89:148–51. [PMC free article: PMC1719787] [PubMed: 14736631]
- Smith CA, Driscoll DA, Emanuel BS, McDonald-McGinn DM, Zackai EH, Sullivan KE. Increased prevalence of immunoglobulin A deficiency in patients with the chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Diagn Lab Immunol. 1998;5:415-7. [PMC free article: PMC104536] [PubMed: 9606003]
- Sullivan KE, McDonald-McGinn D, Driscoll DA, Emanuel BS, Zackai EH, Jawad AF. Longitudinal analysis of lymphocyte function and numbers in the first year of life in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Diagn Lab Immunol. 1999;6:906-11. [PMC free article: PMC95796] [PubMed: 10548584]
- Sullivan KE, McDonald-McGinn DM, Driscoll DA, Zmijewski CM, Ellabban AS, Reed L, Emanuel BS, Zackai EH, Athreya BH, Keenan G. Juvenile rheumatoid arthritis-like polyarthritis in chromosome 22q11.2 deletion syndrome (DiGeorge anomalad/velocardiofacial syndrome/conotruncal anomaly face syndrome). Arthritis Rheum. 1997;40:430-6. [PubMed: 9082929]
- Sullivan KE. Chromosome 22q11.2 deletion syndrome and DiGeorge syndrome. Immunol Rev. 2019;287:186-201. [PubMed: 30565249]
- Swillen A, McDonald-McGinn D. Developmental trajectories in 22q11.2 deletion. Am J Med Genet C Semin Med Genet. 2015;169:172-81. [PMC free article: PMC5061035] [PubMed: 25989227]
- Swillen A, Moss E, Duijff S. Neurodevelopmental outcome in 22q11.2 deletion syndrome and management. Am J Med Genet A. 2018;176:2160-6. [PMC free article: PMC6202262] [PubMed: 29696780]
- Villalón-Reina JE, Martínez K, Qu X, Ching CRK, Nir TM, Kothapalli D, Corbin C, Sun D, Lin A, Forsyth JK, Kushan L, Vajdi A, Jalbrzikowski M, Hansen L, Jonas RK, van Amelsvoort T, Bakker G, Kates WR, Antshel KM, Fremont W, Campbell LE, McCabe KL, Daly E, Gudbrandsen M, Murphy CM, Murphy D, Craig M, Emanuel B, McDonald-McGinn DM, Vorstman JAS, Fiksinski AM, Koops S, Ruparel K, Roalf D, Gur RE, Eric Schmitt J, Simon TJ, Goodrich-Hunsaker NJ, Durdle CA, Doherty JL, Cunningham AC, van den Bree M, Linden DEJ, Owen M, Moss H, Kelly S, Donohoe G, Murphy KC, Arango C, Jahanshad N, Thompson PM, Bearden CE. Altered white matter microstructure in 22q11.2 deletion syndrome: a multisite diffusion tensor imaging study. Mol Psychiatry. 2020;25:2818-31. [PMC free article: PMC6986984] [PubMed: 31358905]
- Weinzimer SA, McDonald-McGinn DM, Driscoll DA, Emanuel BS, Zackai EH, Moshang T Jr. Growth hormone deficiency in patients with 22q11.2 deletion: expanding the phenotype. Pediatrics. 1998;101:929-32. [PubMed: 9565428]
- Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366-73. [PubMed: 14585638]
- Yamagishi H, Garg V, Matsuoka R, Thomas T, Srivastava D. A molecular pathway revealing a genetic basis for human cardiac and craniofacial defects. Science. 1999;283:1158-61. [PubMed: 10024240]
Publication Details
Author Information and Affiliations
Philadelphia, Pennsylvania
Philadelphia, Pennsylvania
Children's Hospital of Philadelphia
Philadelphia, Pennsylvania
Philadelphia, Pennsylvania
Publication History
Initial Posting: September 23, 1999; Last Revision: May 9, 2024.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
McDonald-McGinn DM, Hain HS, Emanuel BS, et al. 22q11.2 Deletion Syndrome. 1999 Sep 23 [Updated 2024 May 9]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.