Summary
Clinical characteristics.
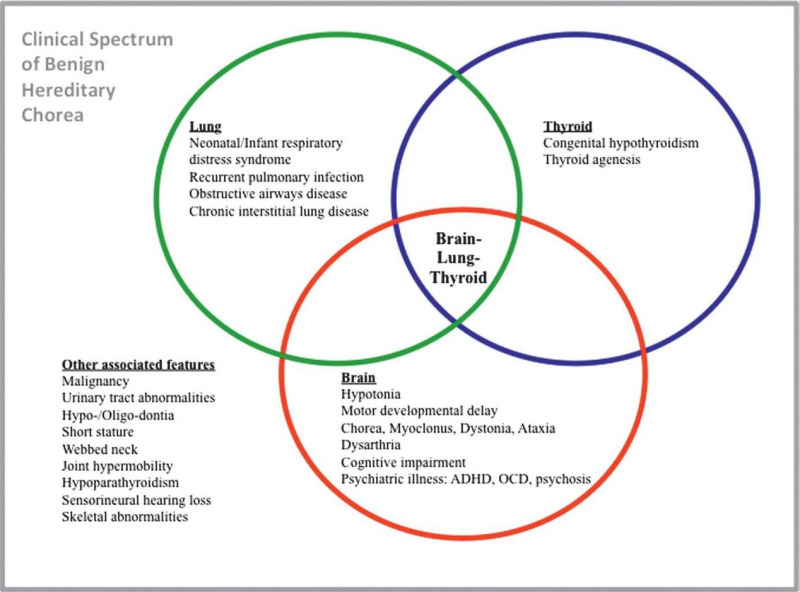
NKX2-1-related disorders range from benign hereditary chorea (BHC) to choreoathetosis, congenital hypothyroidism, and neonatal respiratory distress syndrome (also known as brain-lung-thyroid syndrome). Childhood-onset chorea, the hallmark feature of NKX2-1-related disorders, may or may not be associated with pulmonary disease or congenital hypothyroidism. Age of onset of chorea varies from early infancy (most commonly) to late childhood or adolescence and may progress into the second decade, after which it remains static or (rarely) remits. Pulmonary disease, the second most common manifestation, can include respiratory distress syndrome in neonates, interstitial lung disease in young children, and pulmonary fibrosis in older individuals. The risk for pulmonary carcinoma is increased in young adults with NKX2-1-related disorders. Thyroid dysfunction, occurring as a result of thyroid dysgenesis, can present as congenital or compensated hypothyroidism. In one review, 50% of affected individuals had the full brain-lung-thyroid syndrome, 30% had brain and thyroid involvement only, and 13% had chorea only.
Diagnosis/testing.
The diagnosis of NKX2-1-related disorders is established in a proband with a heterozygous pathogenic variant in NKX2-1 identified by molecular genetic testing.
Management.
Treatment of manifestations: Tetrabenazine, deutetrabenazine, or valbenazine starting at low doses and gradually increasing to control chorea. Levodopa, reported to improve chorea in children with gait abnormalities (including frequent falls), can be considered as second-line therapy for the treatment of chorea and as first-line therapy in children with gait impairment. Pulmonary manifestations are treated per standard recommendations including respiratory support as needed during the neonatal period, RSV vaccination in infancy, and early treatment of asthma and interstitial lung disease. Standard treatment for pulmonary carcinoma. Thyroid replacement therapy for hypothyroidism. Early intervention and physical therapy for motor and gait abnormalities. Standard treatments for neuropsychiatric issues.
Surveillance: Annual neurologic evaluation or more frequently as needed, depending on symptoms. Annual pulmonary evaluations including pulmonary function tests and chest x-ray or chest CT scan to screen for pulmonary malignancy. Annual endocrine evaluations including thyroid function tests (serum thyroxine and thyroid-stimulating hormone) and physical examination (including thyroid palpation) to screen for thyroid cancer.
Agents/circumstances to avoid: Dopamine receptor blockers due to the risk of developing tardive dyskinesia, which can worsen choreiform movements.
Evaluation of relatives at risk: Testing at-risk relatives prenatally or as soon as possible after birth for early identification and treatment of infants at risk for congenital hypothyroidism (and associated neurodevelopmental consequences) as well as pulmonary disease.
Pregnancy management: Prior to pregnancy or early in gestation, assess the safety of medications used for the treatment of chorea.
Genetic counseling.
NKX2-1-related disorders are inherited in an autosomal dominant manner. Most individuals with an NKX2-1-related disorder have an affected parent. Each child of an individual with an NKX2-1-related disorder has a 50% chance of inheriting the pathogenic variant. Once the NKX2-1 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk is possible.
GeneReview Scope
Table
Benign hereditary chorea (BHC) Choreoathetosis, congenital hypothyroidism, and neonatal respiratory distress syndrome (collectively also known as brain-lung-thyroid syndrome)
Diagnosis
Suggestive Findings
NKX2-1-related disorders should be suspected in probands with the following clinical and laboratory findings and family history.
Clinical findings
- Infancy- or childhood-onset non-progressive chorea that may or may not be associated with:
- Mild neurologic symptoms including axial hypotonia, ataxia, developmental delays, impaired mobility of upper and lower limbs;
- Congenital hypothyroidism;
- Respiratory distress syndrome;
OR - A history of congenital hypothyroidism and:
- Neurologic manifestations including hypotonia, neurodevelopmental delay, seizures; and/or
- Respiratory dysfunction including interstitial lung disease in children.
Laboratory findings
- Congenital hypothyroidism (i.e., low thyroid hormone production with elevated thyroid-stimulating hormone)
- Compensated hypothyroidism (i.e., low-to-normal thyroid hormone production with elevated thyroid-stimulating hormone)
Family history is consistent with autosomal dominant inheritance (e.g., affected males and females in multiple generations). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of an NKX2-1-related disorder is established in a proband with suggestive findings and a heterozygous pathogenic (or likely pathogenic) variant in NKX2-1 identified by molecular genetic testing [Inzelberg et al 2011] (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Therefore, reference to "pathogenic variant" in this section is understood to include any likely pathogenic variant. (2) Identification of a heterozygous NKX2-1 variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (chromosomal microarray analysis, exome sequencing, genome sequencing). Gene-targeted testing requires that the clinician determine which gene(s) are likely involved (see Option 1), whereas genomic testing does not (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of an NKX2-1-related disorder, molecular genetic testing approaches can include single-gene testing or a multigene panel.
- Single-gene testing. Sequence analysis of NKX2-1 is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
- A multigene panel that includes NKX2-1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the diagnosis of an NKX2-1-related disorder has not been considered because an individual has atypical phenotypic features, comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible. To date, the majority of reported NKX2-1 pathogenic variants are within the coding region and are likely to be identified on exome sequencing.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Chromosomal microarray analysis (CMA) uses oligonucleotide or SNP arrays to detect genome-wide large deletions/duplications (including NKX2-1) that cannot be detected by sequence analysis (see Table 1). CMA is recommended for the evaluation of childhood-onset chorea if multigene panel and comprehensive genomic testing are negative.
For an introduction to CMA click here. More detailed information for clinicians ordering genetic tests can be found here.
Table 1.
Molecular Genetic Testing Used in NKX2-1-Related Disorders
Clinical Characteristics
Clinical Description
NKX2-1-related disorders include benign hereditary chorea (BHC) and choreoathetosis, congenital hypothyroidism, and neonatal respiratory distress syndrome (collectively known as brain-lung-thyroid syndrome). Individuals can have one or more of several features associated with this spectrum (see Figure 1). To date, more than 120 individuals have been identified with a pathogenic variant in NKX2-1 [Gras et al 2012, Peall & Kurian 2015, Kharbanda et al 2017]. In a review of 46 affected individuals, Carré et al [2009] found that 50% had the full brain-lung-thyroid syndrome, 30% had only brain and thyroid involvement, and 13% had isolated chorea. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
NKX2-1-Related Disorders: Frequency of Select Features
Neurologic Manifestations
Chorea, an involuntary, random, irregular, jerk-like, and continuous movement, is a classic early finding in BHC and other NKX2-1-related disorders. The onset of chorea generally occurs during one of the following time periods:
- Early infancy or within the first year of life (most common)
- Late childhood or adolescence
Chorea progresses into the second decade, after which it remains static or may spontaneously remit [Kleiner-Fisman et al 2003, Peall & Kurian 2015]. Although originally referred to as "benign" hereditary chorea, the neurologic manifestations of NKX2-1-related disorders can be quite disabling, due to chorea as well as gait and balance abnormalities, hypotonia, and a variety of other motor and non-motor manifestations [Parnes et al 2018].
Chorea typically involves all body regions (e.g., face, tongue, neck, trunk, limbs) and may be associated with motor and gait abnormalities, possibly secondary to the choreiform movements. The movements are jerk-like and spread randomly from one body part to another; they often worsen with stress and may disappear with sleep. Children with BHC may fall frequently [Kleiner-Fisman et al 2003]. Although affected children may be delayed in walking, persistent gait abnormalities are rare [McMichael et al 2013]. Rosati et al [2015] described two unrelated children presenting with spontaneous falls without loss of consciousness preceding the development of chorea.
The prevalence of chorea in NKX2-1-related disorders is unknown. In one study, all 28 affected individuals from 13 families with a heterozygous NKX2-1 pathogenic variant had chorea and hypotonia [Gras et al 2012]. However, in a retrospective study of 21 individuals with NKX2-1-related disorders presenting with pulmonary dysfunction, at least two unrelated individuals and three members of one family did not have any neurologic symptoms [Hamvas et al 2013].
Other less common neurologic manifestations
- Hypotonia, incoordination, and motor delays, which are common during the neonatal period / infancy but rarely reported in childhood/adulthood [Peall et al 2014, Veneziano et al 2014]
- Myoclonus, dystonia, and/or ataxia [Peall et al 2014, de Gusmao et al 2015]
- Intention tremor [Peall et al 2014]
- Dysarthria [Peall et al 2014]
- Restless leg syndrome [Iodice et al 2019]
- Neurocognitive changes, including learning difficulties and reduced working memory and attention [Gras et al 2012, Peall et al 2014, Parnes et al 2018, Graziola et al 2021]
- Seizures [Villamil-Osorio et al 2021]
In one report, two sibs initially diagnosed with ataxic dyskinetic cerebral palsy were later found to have a pathogenic variant in NKX2-1 [McMichael et al 2013].
Neuropsychiatric manifestations including attention-deficit/hyperactivity disorder have been reported [Gras et al 2012]. Although psychiatric disorders are rare in individuals with an NKX2-1 pathogenic variant, schizophrenia [Glik et al 2008], postpartum psychosis [Salvatore et al 2010], and obsessive-compulsive disorder [Peall et al 2014] have been reported.
Neuroimaging is usually normal. Structural brain abnormalities have been rarely reported, including abnormal sella turcica [Krude et al 2002, Balicza et al 2018], agenesis of the corpus callosum [Carré et al 2009], cavum septum pellucidum, and microcephaly [Iwatani et al 2000]. Hypoplastic pallidum and lack of differentiation of medial and lateral components of the pallidum were reported in a single individual, and bilateral pallidal signal hyperintensities on T2-weighted MRI were described in another individual [Kleiner-Fisman & Lang 2007]. An expanding pituitary cyst was reported in two related individuals with a novel NKX2-1 pathogenic variant [Veneziano et al 2014]. Chiari I malformation was also reported in one female at age 24 months [Gonçalves et al 2019].
Single-photon emission computed tomography (SPECT) has demonstrated reduced cerebral blood flow to the basal ganglia bilaterally, and more specifically to the caudate nuclei, in three affected individuals [Uematsu et al 2012]. Subtle abnormalities in presynaptic dopamine transporter function utilizing positron emission tomography (PET) imaging have also been reported in two affected individuals [Konishi et al 2013].
Neuropathology. Autopsies of two individuals with NKX2-1-related disorders did not identify gross or microscopic abnormalities of the brain, but showed reduced number of striatal and neocortical interneurons consistent with a defect in neuronal migration and supporting the theory that these disorders are due to abnormalities in brain development rather than neurodegeneration [Kleiner-Fisman et al 2003].
Pulmonary Manifestations
Pulmonary dysfunction is the second most common manifestation of NKX2-1-related disorders. In a meta-analysis of 29 published reports of NKX2-1-related disorders, up to 49% (61/124) of affected individuals had pulmonary manifestations of varying severity [Gras et al 2012]. Clinical presentation and course vary among affected individuals.
- Respiratory distress syndrome (RDS) with or without pulmonary hypertension is most common in the neonatal period.
- Neuroendocrine cell hyperplasia, a distinct form of childhood interstitial lung disease (ILD), can be present in infancy; it typically improves with age [Young et al 2013]. ILD can occur between the ages of four months to seven years.
- Pulmonary fibrosis can occur in older individuals [Hamvas et al 2013].
The highest risk for respiratory distress is in the neonatal period. Affected infants often require mechanical ventilation [Carré et al 2009]. Although usually not fatal, NKX2-1-related disorders have resulted in death from respiratory failure in three infants in the immediate postnatal period to date [Maquet et al 2009, Kleinlein et al 2011, Gillett et al 2013]. However, in most infants, respiratory function typically improves over time [Young et al 2013].
As a result of pulmonary involvement, individuals with NKX2-1-related disorders are at increased risk for recurrent pulmonary infections and chronic interstitial lung disease [Carré et al 2009, Inzelberg et al 2011, Peca et al 2011]. High-resolution CT scan of six children with pathogenic variants in NKX2-1 identified ground-glass opacities and pulmonary consolidation [LeMoine et al 2019].
Pulmonary histologic abnormalities. In one restrospective study of individuals with known pulmonary dysfunction and a pathogenic variant in NKX2-1, histologic abnormalities included interstitial widening and pneumocyte hyperplasia, desquamative interstitial pneumonia, accumulation of foamy alveolar macrophages, and pulmonary alveolar proteinosis [Hamvas et al 2013].
Pulmonary carcinoma. The risk for pulmonary carcinoma is increased in young adults (early 20s) with NKX2-1-related disorders [Fernandez et al 2001, Willemsen et al 2005, Glik et al 2008]. Screening for pulmonary carcinoma in adolecence is recommended (see Surveillance).
Thyroid Manifestations
Thyroid dysfunction, which results from thyroid dysgenesis, can present as congenital hypothyroidism (i.e., reduced or absent production of thyroid hormone) or compensated hypothyroidism (i.e., low-to-normal thyroid hormone production with elevated thyroid-stimulating hormone) [Moya et al 2006, Montanelli & Tonacchera 2010, Gras et al 2012]. Of note, thyroid dysgenesis can manifest structurally as thyroid hypoplasia or hemiagenesis (11/31) or complete absence of the thyroid gland (3/31) [Carré et al 2009].
Thyroid dysfunction varies between individuals with NKX2-1-related disorders and within families of multiple affected individuals. In a meta-analysis of 46 individuals reported with NKX2-1-related disorders, 40 had documented overt or subclinical hypothyroidism, and only six had normal thyroid function [Carré et al 2009].
Currently, newborn screening for hypothyroidism is available in most countries and includes measuring levels of thyroid-stimulating hormone with or without measuring thyroxine levels. Of note, congenital hypothyroidism can be the only manifestation of an NKX2-1-related disorder.
Other Features
Other features reported in single individuals or families include the following:
- Hypo- or oligodontia [Devos et al 2006, Guala et al 2008]
- Urinary tract abnormalities:
- Vesicoureteral reflux [Ferrara et al 2008, Salvatore et al 2010]
- Megacystis [Ferrara et al 2008, Salvatore et al 2010]
- Endocrine abnormalities:
- Hypogonadotropic hypogonadism [Balicza et al 2018]
- Growth hormone deficiency [Balicza et al 2018, Trevisani et al 2021]
- Short stature (without documented growth hormone deficiency) [Glik et al 2008]
- Webbed neck [Thorwarth et al 2014]
- Cardiac abnormalities:
- Structural cardiac defects (ventricular or atrial septal defects) [Thorwarth et al 2014]
- Patent foramen ovale [Ferrara et al 2008, Salvatore et al 2010]
- Joint laxity [Parnes et al 2018]
- Pes cavus [Peall et al 2014]
- Kyphosis [Peall et al 2014]
Prognosis and Progression
Life expectancy in individuals with NKX2-1-related disorders is expected to be normal [Fernandez et al 2001].
A retrospective study describing 28 individuals with 13 novel NKX2-1 pathogenic variants over a mean duration of 24.5 years reported a homogeneous progression of neurologic manifestations. Hypotonia is present in the first year of life with or without delays in motor milestones or early chorea. Chorea is generally mild and improves until puberty through early adulthood, when it typically stabilizes. In some individuals, it resolves entirely in adulthood [Gras et al 2012].
There is limited information on the long-term prognosis of pulmonary and thyroid manifestations. Progression is rare and features may improve in adulthood [Gras et al 2012]. Individuals with lung involvement are at risk of respiratory failure in early infancy, as well as recurrent infections and asthma throughout life. Compensated hypothyroidism is typically well controlled in individuals with NKX2-1-related disorders.
Genotype-Phenotype Correlations
Manifestations of NKX2-1-related disorders vary among individuals even within the same family.
Missense NKX2-1 variants have been associated with milder phenotypes, though a systematic analysis of published reports revealed no clear correlation between variant type and protein function [Gras et al 2012, Monti et al 2015].
Penetrance
No studies have evaluated the penetrance of NKX2-1 pathogenic variants to date. Published case series provide limited descriptions of penetrance across families, though intrafamilial variation has been reported.
Nomenclature
Before the molecular basis was known, the disorder now known to be caused by a heterozygous pathogenic variant in NKX2-1 [Inzelberg et al 2011] was referred to as benign hereditary chorea (BHC) based on the original description of non-progressive familial chorea in a five-generation family [Haerer et al 1967]. The broad phenotypic spectrum associated with pathogenic variants in NKX2-1 (including BHC and a variable combination of lung, thyroid, and neurologic abnormalities) led Willemsen et al [2005] to coin the term "brain-lung-thyroid syndrome." Considering the variable manifestations of individuals with pathogenic variants in NKX2-1, the authors suggest that these disorders be referred to as NKX2-1-related disorders.
Of note, NKX2-1 was previously known as TITF-1; thus, early literature describing the molecular basis of this disorder uses this gene designation [Breedveld et al 2002, Kleiner-Fisman et al 2003, Costa et al 2005, Devos et al 2006, Kleiner-Fisman & Lang 2007, Glik et al 2008].
Prevalence
More than 120 individuals with NKX2-1-related disorders have been described in the literature. The prevalence of NKX2-1-related disorders is unknown.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with pathogenic variants in NKX2-1.
Somatic variants in NKX2-1 have been identified in sporadic papillary thyroid and sporadic pulmonary carcinomas (lung adenocarcinoma, non-small cell lung cancer) in individuals without any other findings of NKX2-1-related disorders [Ngan et al 2009, Matsuse et al 2011, Watanabe et al 2013, Tsai et al 2014]. In these instances, the NKX2-1 variant is not present in the germline and therefore predisposition to these tumors is not heritable.
Differential Diagnosis
The differential diagnosis of NKX2-1-related disorders includes hereditary (see Table 3a) and acquired (see Table 3b) causes of chorea, congenital hypothyroidism, and respiratory insufficiency.
Table 3a.
Selected Hereditary Disorders in the Differential Diagnosis of NKX2-1-Related Disorders
Other hereditary disorders associated with chorea that can be considered in the differential diagnosis include:
- Neurometabolic disorders (Lesch-Nyhan disease, lysosomal storage disorders, amino acid deficiency disorders, Leigh syndrome)
Table 3b.
Acquired Disorders in the Differential Diagnosis of NKX2-1-Related Disorders
Management
No clinical practice guidelines for NKX2-1-related disorders have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with an NKX2-1-related disorder, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
NKX2-1-Related Disorders: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
Supportive care to improve the quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 5).
Table 5.
NKX2-1-Related Disorders: Treatment of Manifestations
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 6 are recommended.
Table 6.
NKX2-1-Related Disorders: Recommended Surveillance
Agents/Circumstances to Avoid
Although Glik et al [2008] reported amelioration of choreic movements in an individual with benign hereditary chorea treated with olanzapine for psychosis, dopamine receptor blockers are not recommended as a first-line therapy for the treatment of chorea. Olanzapine and dopamine receptor blockers, while effective in treating choreiform movements, are associated with a small risk of developing tardive dyskinesia, which can worsen choreiform movements long term.
Due to the risk of pulmonary disease in NKX2-1-related disorders, exposure to smoking may exacerbate pulmonary dysfunction and increase the risk of lung cancer in affected individuals.
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of at-risk relatives prenatally or as soon as possible after birth to identify infants at high risk for congenital hypothyroidism and pulmonary disease, establish the diagnosis and initiate medical management early, and, importantly, prevent the neurodevelopmental consequences of untreated congenital hypothyroidism. In most countries, newborn screening for congenital hypothyroidism is performed; however, if this is not routine practice, it is recommended to screen at-risk newborns for congenital hypothyroidism.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
There is no known increased risk during pregnancy for a woman with an NKX2-1-related disorder.
Prior to pregnancy or early in gestation, it is recommended that a woman work with her physician to determine whether any medication she is taking for chorea is safe for the fetus.
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
NKX2-1-related disorders are inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- Most individuals diagnosed with an NKX2-1-related disorder have an affected parent.
- A proband with an NKX2-1-related disorder may have the disorder as the result of a de novo pathogenic variant; however, the proportion of probands who have a de novo pathogenic variant is unknown.
- If the proband appears to be the only affected family member (i.e., a simplex case), molecular genetic testing is recommended for the parents of the proband to confirm their genetic status and to allow reliable recurrence risk counseling.
- If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Note: Testing parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.
- The family history of some individuals diagnosed with an NKX2-1-related disorder may appear to be negative because of failure to recognize the disorder in family members due to mild manifestations, early death of the parent before the onset of symptoms, or late onset of the disease in the affected parent. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has demonstrated that neither parent is heterozygous for the pathogenic variant identified in the proband.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%.
- If the NKX2-1 pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretical possibility of parental germline mosaicism [Rahbari et al 2016].
- If the parents have not been tested for the NKX2-1 pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for an NKX2-1-related disorder because of the possibility of reduced penetrance in a heterozygous parent or the theoretical possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with an NKX2-1-related disorder has a 50% chance of inheriting the pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the NKX2-1 pathogenic variant, the parent's family members may be at risk.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Predictive testing for at-risk asymptomatic family members requires prior identification of the pathogenic variant in the family.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the NKX2-1 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- Newborn Screening in Your StateHealth Resources & Services Administration
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
NKX2-1-Related Disorders: Genes and Databases
Table B.
OMIM Entries for NKX2-1-Related Disorders (View All in OMIM)
NKX2-1 encodes the homeobox protein Nkx-2.1. This protein, also known as thyroid transcription factor 1 (TTF-1), plays a critical role during organogenesis of the basal ganglia, lungs, and thyroid [Guillot et al 2010]. In vivo studies have shown that NKX2-1 is implicated in the development of the globus pallidus and striatal cholinergic neurons [Flandin et al 2010], as well as development of the lungs, surfactant production, and maintenance of surfactant homeostasis [Kleinlein et al 2011, Peca et al 2011]. Supporting the role of NKX2-1 in lung development, pathogenic variants in NKX2-1 are commonly associated with impaired pulmonary branching and reduced alveolar counts [Galambos et al 2010]. NKX2-1 is also important for early thyroid development in addition to maintenance of the architecture and function of the fully developed thyroid gland [Kusakabe et al 2006, Fagman & Nilsson 2011, Kimura 2011].
Mechanism of disease causation. Loss of function (due to haploinsufficiency)
NKX2-1-specific laboratory technical considerations. In individuals without a pathogenic variant identified by NKX2-1 sequencing and deletion/duplication analysis, chromosomal microarray should be performed, as large deletions downstream of NKX2-1 have been described [Villamil-Osorio et al 2021].
There are several NKX2-1 transcripts. The longer transcript (NM_001079668.2) comprises three exons. Most pathogenic variants in NKX2-1 have been reported in this transcript. A shorter transcript has been identified (NM_003317.3).
Chapter Notes
Author Notes
USH University Medical Center Parkinson's Disease and Movement Disorders Care
Revision History
- 29 June 2023 (gm) Comprehensive update posted live
- 29 July 2016 (ha) Comprehensive updated posted live
- 20 February 2014 (me) Review posted live
- 9 September 2013 (np) Original submission
References
Literature Cited
- Asmus F, Horber V, Pohlenz J, Schwabe, D, Zimprich A, Munz M, Schöning M, Gasser T. A novel TITF-1 mutation causes benign hereditary chorea with response to levodopa. Neurology 2005;64:1952-4 [PubMed: 15955952]
- Balicza P, Grosz Z, Molnár V, Illés A, Csabán D, Gézsi A, Dézsi L, Zádori D, Vécsei L, Molnár MJ. NKX2-1 new mutation associated with myoclonus, dystonia, and pituitary involvement. Front Genet. 2018;9:335. [PMC free article: PMC6113386] [PubMed: 30186310]
- Barnett CP, Mencel JJ, Gecz J, Waters W, Kirwin SM, Vinette KM, Uppill M, Nicholl J. Choreoathetosis, congenital hypothyroidism and neonatal respiratory distress syndrome with intact NKX2-1. Am J Med Genet A. 2012;158A:3168-73. [PubMed: 23169673]
- Bashir H, Jankovic J. Treatment options for chorea. Expert Rev Neurother. 2018;18:51-63. [PubMed: 29120264]
- Breedveld GJ, van Dongen JW, Danesino C, Guala A, Percy AK, Dure LS, Harper P, Lazarou LP, van der Linde H, Joosse M, Grüters A, MacDonald ME, de Vries BB, Arts WF, Oostra BA, Krude H, Heutink P. Mutations in TITF-1 are associated with benign hereditary chorea. Hum Mol Genet 2002;11:971-9. [PubMed: 11971878]
- Carré A, Szinnai G, Castanet M, Sura-Trueba S, Tron E, Broutin-L'Hermite I, Barat P, Goizet C, Lacombe D, Moutard ML, Raybaud C, Raynaud-Ravni C, Romana S, Ythier H, Léger J, Polak M. Five new TTF1/NKX2.1 mutations in brain-lung-thyroid syndrome: rescue by PAX8 synergism in one case. Hum Mol Genet. 2009;18:2266-76. [PubMed: 19336474]
- Costa MC, Costa C, Silva AP, Evangelista P, Santos L, Ferro A, Segeiros J, Maciel P. Nonsense mutation in TITF1 in a Portuguese family with benign hereditary chorea. Neurogenetics. 2005;6:209-15. [PubMed: 16220345]
- de Gusmao CM, Kok F, Casella EB, Waugh JL. Benign hereditary chorea related to NKX2-1 with ataxia and dystonia. Neurol Genet. 2015;2:e40. [PMC free article: PMC4817908] [PubMed: 27066577]
- Devos D, Vuillaume I, de Becdelievre A, de Martinville B, Dhaenens CM, Cuvellier JC, Cuisset JM, Vallee L, Lemaitre MP, Bourteel H, Hachulla E, Wallaert B, Destee A, Defebvre L, Sablonniere B. New syndromic form of benign hereditary chorea is associated with a deletion of TITF-1 and PAX-9 contiguous genes. Mov Disord. 2006;21:2237-40. [PubMed: 17044090]
- Fagman H, Nilsson M. Morphogenetics of early thyroid development. J Mol Endocrinol. 2011; 46:R33-42. [PubMed: 21322126]
- Fernandez M, Raskind W, Matsushita M, Wolff J, Lipe H, Bird T. Hereditary benign chorea: clinical and genetic features of a distinct disease. Neurology. 2001;57:106-10. [PubMed: 11445636]
- Ferrara AM, De Michele G, Salvatore E, Di Maio L, Zampella E, Capuano S, Del Prete G, Fenzi G, Filla A, Macchia PE. A novel NKX2.1 mutation in a family with hypothyroidism and benign hereditary chorea. Thyroid. 2008;18:1005-9. [PubMed: 18788921]
- Flandin P, Kimura S, Rubenstein JL. The progenitor zone of the ventral medial ganglionic eminence requires Nkx2-1 to generate most of the globus pallidus but few neocortical interneurons. J Neurosci. 2010;30:2812-23. [PMC free article: PMC2865856] [PubMed: 20181579]
- Galambos C, Levy H, Cannon CL, Vargas SO, Reid LM, Cleveland R, Lindeman R, deMello DE, Wert SE, Whitsett JA, Perez-Atayde AR, Kozakewich H. Pulmonary pathology in thyroid transcription factor-1 deficiency syndrome. Am J Respir Crit Care Med. 2010;182:549-54. [PMC free article: PMC2937244] [PubMed: 20203240]
- Gillett ES, Deutsch GH, Bamshad MJ, McAdams RM, Mann PC. Novel NKX2.1 mutation associated with hypothyroidism and lethal respiratory failure in a full-term neonate. J Perinatol. 2013;33:157-60. [PubMed: 23361500]
- Glik A, Vuillaume I, Devos D, Inzelberg R. Psychosis, short stature in benign hereditary chorea: a novel thyroid transcription factor-1 mutation. Mov Disord. 2008;23:1744-7. [PubMed: 18661567]
- Gonçalves D, Lourenço L, Guardiano M, Castro-Correia C, Sampaio M, Leão M. Chiari malformation type I in a patient with a novel NKX2-1 mutation. J Pediatr Neurosci. 2019;14:169-72. [PMC free article: PMC6798275] [PubMed: 31649781]
- Gras D, Jonard L, Roze E, Chantot-Bastaraud S, Koht J, Motte J, Rodriguez D, Louha M, Caubel I, Kemlin I, Lion-Francois L, Goizet C, Guillot L, Moutard ML, Epaud R, Heron B, Charles P, Tallot M, Camuzat A, Durr A, Polak A, Devos D, Sanlaville D, Vuillaume I, Billette de Villemeur T, Vidailhet M, Doummar D. Benign hereditary chorea: phenotype, prognosis, therapeutic outcome and long term follow-up in a large series with new mutations in the TITF1/NKX2-1 gene. J Neurol Neurosurg Psychiatry. 2012;83:956-62. [PubMed: 22832740]
- Graziola F, Garone G, Grasso M, Schirinzi T, Capuano A. Working memory, attention and planning abilities in NKX2.1-related chorea. Parkinsonism Relat Disord. 2021;88:24-7. [PubMed: 34091414]
- Guala A, Falco V, Breedveld G, De Filippi P, Danesino C. Deletion of PAX9 and oligodontia: a third family and review of the literature. Int J Paediatr Dent. 2008;18:441-5. [PubMed: 18445003]
- Guillot L, Carre A, Szinnai G, Castanet M, Tron E, Jaubert F, Broutin I, Counil F, Feldmann D, Clement A, Polak M, Epaud R. NKX2-1 mutations leading to surfactant protein promoter dysregulation cause interstitial lung disease in "brain-lung-thyroid syndrome". Hum Mutat. 2010;31:E1146-62. [PubMed: 20020530]
- Haerer AF, Currier RD, Jackson JF. Hereditary nonprogressive chorea of early onset. N Engl J Med. 1967;276:1220-4. [PubMed: 4225827]
- Hamvas A, Deterding RR, Wert SE, White FV, Dishop MK, Alfano DN, Halnower A, Planer B, Stephan MJ, Uchinda DA, Williames LD, Rosenfeld JA, Lebel RR, Young LR, Cole FS, Nogee LM. Heterogeneous pulmonary phenotypes associated with mutations in the thyroid transcription factor gene NKX2-1. Chest. 2013;144:794-804. [PMC free article: PMC3760742] [PubMed: 23430038]
- Invernizzi F, Zorzi G, Legati A, Coppola G, D'Adamo P, Nardocci N, Garavaglia B, Ghezzi D. Benign hereditary chorea and deletions outside NKX2-1: what's the role of MBIP? Eur J Med Genet. 2018;61:581-4. [PubMed: 29621620]
- Inzelberg R, Weinberger M, Gak E. Benign hereditary chorea: an update. Parkinsonism Relat Disord. 2011;17:301-7. [PubMed: 21292530]
- Iodice A, Carecchio M, Zorzi G, Garavaglia B, Spagnoli C, Salerno GG, Frattini D, Mencacci NE, Invernizzi F, Veneziano L, Mantuano E, Angriman M, Fusco C. Restless legs syndrome in NKX2-1-related chorea: an expansion of the disease spectrum. Brain Dev. 2019;41:250-6. [PubMed: 30352709]
- Iwatani N, Mabe H, Devriendt K, Kodama M, Miike T. Deletion of NKX2.1 gene encoding thyroid transcription factor-1 in two siblings with hypothyroidism and respiratory failure. J Pediatr. 2000;137:272-6. [PubMed: 10931427]
- Jankovic J. Treatment of hyperkinetic movement disorders. Lancet Neurol 2009;8:844-56. [PubMed: 19679276]
- Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother 2011;11:1509-23. [PubMed: 22014129]
- Jimenez-Shahed J, Jankovic J. Tetrabenazine for treatment of chorea associated with Huntington’s disease. Expert Opin Orphan Drugs 2013;1:423-36.
- Kharbanda M, Hermanns P, Jones J, Pohlenz J, Horrocks I, Donaldson M. A further case of brain-lung-thyroid syndrome with deletion proximal to NKX2-1. Eur J Med Genet. 2017;60:257-60. [PubMed: 28286255]
- Kimura S. Thyroid-specific transcription factors and their roles in thyroid cancer. J Thyroid Res. 2011;2011:710213. [PMC free article: PMC3112524] [PubMed: 21687604]
- Kleiner-Fisman G, Lang AE. Benign hereditary chorea revisited: a journey to understanding. Mov Disord 2007;22:2297-305. [PubMed: 17702033]
- Kleiner-Fisman G, Rogaeva E, Halliday W, Houle S, Kawarai T, Sato C, Medeiros H, St George-Hyslop PH, Lang AE. Benign hereditary chorea: clinical, genetic, and pathological findings. Ann Neurol 2003;54:244-7. [PubMed: 12891678]
- Kleinlein B, Griese M, Liebisch G, Krude H, Lohse P, Aslanidis C, Schmitz G, Pters J, Holzinger A. Fatal neonatal respiratory failure in an infant with congenital hypothyroidism due to haploinsufficiency of the NKX2-1 gene: alteration of pulmonary surfactant homeostasis. Arch Dis Child Fetal Neonatal Ed. 2011;96:F453-6. [PubMed: 20584796]
- Konishi T, Kono S, Fujimoto M, Terada T, Matsushita K, Ouchi Y, Miyajima H. Benign hereditary chorea: dopaminergic brain imaging in patients with a novel intronic NKX2.1 gene mutation. J Neurol. 2013;260:207-13. [PubMed: 22825795]
- Krude H, Schutz B, Biebermann H, von Moers A, Schnabel D, Neitzel H, Tonnies H, Weise D, Lafferty A, Schwarz S, Defelice M, Von Deimling A, Van Langeghem F, Dilauro R, Gruters A. Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2-1 haploinsufficiency. J Clin Invest. 2002;109:475-80. [PMC free article: PMC150790] [PubMed: 11854319]
- Kusakabe T, Kawaguchi A, Hoshi N, Kawaguchi R, Hoshi S, Kimura S. Thyoid-specific enhancer-binding protein/NKX2.1 is required for the maintenance of ordered architecture and function of the differentiated thyroid. Mol Endocrinol. 2006; 20:1796-809. [PMC free article: PMC2588428] [PubMed: 16601074]
- LeMoine BD, Browne LP, Liptzin DR, Deterding RR, Galambos C, Weinman JP. High-resolution computed tomography findings of thyroid transcription factor 1 deficiency (NKX2-1 mutations). Pediatr Radiol. 2019;49:869-75. [PubMed: 30927038]
- Liao J, Coffman KA, Locker J, Padiath QS, Nmezi B, Filipink RA, Hu J, Sathanoori M, Madan-Khetarpal S, McGuire M, Schreiber A, Moran R, Friedman N, Hoffner L, Rajkovic A, Yatsenko SA, Surti U. Deletion of conserved non-coding sequences downstream from NKX2-1: a novel disease-causing mechanism for benign hereditary chorea. Mol Genet Genomic Med. 2021;9:e1647. [PMC free article: PMC8123744] [PubMed: 33666368]
- Maquet E, Costagliola S, Parma J, Christophe-Hobertus C, Oligny LL, Fournet JC, Robitaille Y, Vuissoz JM, Payot A, Laberge S, Van Vliet G, Deladoey J. Lethal respiratory failure and mild primary hypothyroidism in a term girl with a de novo heterozygous mutation in the TITF1/NKX2.1 gene. J Clin Endocrinol Metab. 2009;94:197-203. [PubMed: 18957494]
- Matsuse M, Takashashi M, Mitsutake N, Nishihara E, Hirokawa M, Kawaguchi T, Rogonovitch T, Saenko V, Bychkov A, Suzuki K, Matsuo K, Tajima K, Miyauchi A, Yamada R, Matsuda F, Yamashita S. The FOXE1 and NKX2-1 loci are associated with susceptibility to papillary thyroid carcinoma in the Japanese population. J Med Genet. 2011;48:645-8. [PubMed: 21730105]
- McMichael G, Haan E, Gardner A, Yap TY, Thompson S, Ouvrier R, Dale RC, Gecz J, Maclennan AH. NKX2-1 mutation in a family diagnosed with ataxic dyskinetic cerebral palsy. Eur J Med Genet. 2013;56:506-9. [PubMed: 23911641]
- Montanelli L, Tonacchera M. Genetics and phenomics of hypothyroidism and thyroid dys- and agenesis due to PAX8 and TTF1 mutations. Mol Cell Endocrinol 2010;322:64-71. [PubMed: 20302910]
- Monti S, Nicoletti A, Cantasano A, Krude H, Cassio A. NKX2.1-related disorders: a novel mutation with mild clinical presentation. Ital J Pediatr. 2015;41:45. [PMC free article: PMC4477322] [PubMed: 26103969]
- Moya CM, Perez de Nanclares G, Castano L, Potau N, Bilbao JR, Carrascosa A, Bargada M, Coya R, Martul P, Vicens-Calvet E, Santisteban P. Functional study of a novel single deletion in the TITF1/NKX2.1 homeobox gene that produces congenital hypothyroidism and benign chorea but not pulmonary distress. J Clin Endocrinol Metab. 2006;91:1832-41. [PubMed: 16507635]
- Ngan ES, Lang BH, Liu T, Shum CK, So MT, Lau DK, Leon TY, Cherny SS, Tsai SY, Lo CY, Khoo US, Tam PK, Garcia-Barceló MM. A germline mutation (A339V) in thyroid transcription factor-1 (TITF-1/NKX2.1) in patients with multinodular goiter and papillary thyroid carcinoma. J Natl Cancer Inst. 2009;101:162-75. [PubMed: 19176457]
- Parnes M, Bashir H, Jankovic J. Is benign hereditary chorea really benign? brain-lung-thyroid syndrome caused by NKX2-1 mutations. Mov Disord Clin Pract. 2018; 6:34-9. [PMC free article: PMC6335533] [PubMed: 30746413]
- Peall KJ, Kurian MA. Benign hereditary chorea: an update. Tremor Other Hyperkinet Mov (N Y). 2015;5:314. [PMC free article: PMC4502401] [PubMed: 26196025]
- Peall KJ, Lumsden D, Kneen R, Madhu R, Peake D, Gibbon F, Lewis H, Hedderly T, Meyer E, Robb SA, Lynch B, King MD, Lin JP, Morris HR, Jungbluth H, Kurian MA. Benign hereditary chorea related to NK2-1: expansion of genotypic and phenotypic spectrum. Dev Med Child Neurol. 2014;56:642-8. [PubMed: 24171694]
- Peca D, Petrini S, Tzialla C, Boldrini R, Morini F, Stronati M, Carnielli VP, Cogo PE, Danhaive O. Altered surfactant homeostasis and recurrent respiratory failure secondary to TTF-1 nuclear targeting defect. Respir Res. 2011;12:115. [PMC free article: PMC3179724] [PubMed: 21867529]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126-33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rosati A, Berti B, Melani F, Cellini E, Procopio E, Guerrini R. Recurrent drop attacks in early childhood as presenting symptom of benign hereditary chorea caused by TITF 1 gene mutations. Dev Med Child Neurol. 2015;57:777-9. [PubMed: 25412988]
- Salvatore E, Di Maio L, Filla A, Ferrara AM, Rinaldi C, Sacca F, Peluso S, Macchia PE, Pappata S, De Michele G, Benign hereditary chorea: clinical and neuroimaging features in an Italian family. Mov Disord 2010;25:1491-6. [PubMed: 20544814]
- Schrag A, Quinn NP, Bhatia KP, Marsden CD. Benign hereditary chorea--entity or syndrome? Mov Disord 2000;15:280-8. [PubMed: 10752577]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197-207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Teissier R, Guillot L, Carre A, Melina M, Stuckens C, Ythier H, Munnich A, Szinnai G, de Blic J, Clement A, Leger J, Castanet M, Epaud R, Polak M. Multiplex ligation-dependent prove amplification improves the detection rate of NKX2.1 mutations in patients affected by brain-lung-thyroid syndrome. Horm Res Paediatr. 2012;77:146-51. [PubMed: 22488412]
- Thorwarth A, Schnitter-Hubener S, Schrumpf P, Muller I, Jyrch S, Dame C, Biebermann H, Katchanov J, Schuelke M, Ebert G, Steininger A, Bonnemann C, Brockmann K, Christen HJ, Crock P, deZegher F, Griese M, Hewitt J, Huber C, Kapelari K, Plecko B, Rating D, Stoeva I, Ropers HH, Gruters A, Ullmann R, Krude H. Comprehensive genotyping and clinical characterization reveals 27 novel KNX2-1 mutations and expand the phenotypic spectrum. J Med Genet. 2014;51:375-87. [PMC free article: PMC5240655] [PubMed: 24714694]
- Trevisani V, Predieri B, Madeo SF, Fusco C, Garavelli L, Caraffi S, Iughetti L. Growth hormone deficiency in a child with benign hereditary chorea caused by a de novo mutation of the TITF1/NKX2-1 gene. J Pediatr Endocrinol Metab. 2021;35:411-5. [PubMed: 34710315]
- Tsai LH, Chen PM, Cheng YW, Chen CY, Sheu GT, Wu TC, Lee H. LKB1 loss by alteration of the NKX2-1/p53 pathway promotes tumor malignancy and predicts poor survival and relapse in lung adenocarcinomas. Oncogene. 2014;33:3851-60. [PubMed: 23995788]
- Uematsu M, Haginoya K, Kikuchi A, Nakayama T, Numata Y, Kobayashi T, Hino-Fukuo N, Fujiwara I, Kure S. Hypoperfusion in caudate nucei in patients with brain-lung-thyroid syndrome. J Neurol Sci. 2012;315:77-81. [PubMed: 22166853]
- Veneziano L, Parkinson MH, Mantuano E, Frontali M, Bhatia KP, Giunti P. A novel de novo mutation of the TITF1/NKX2-1 gene causing ataxia, benign hereditary chorea, hypothyroidism and a pituitary mass in a UK family and review of the literature. Cerebellum. 2014;13:588-95. [PMC free article: PMC4155168] [PubMed: 24930029]
- Villamil-Osorio M, Yunis LK, Quintero L, Restrepo-Gualteros S, Yunis JJ, Jaramillo L, Agudelo BI, Ladino Y. [Brain-lung-thyroid syndrome in a newborn with deletion 14q12-q21.1]. Andes Pediatr. 2021;92:930-6. [PubMed: 35506806]
- Watanabe H, Francis JM, Woo MS, Etemad B, Lin W, Fries DF, Peng S, Snyder EL, Tata PR, Izzo F, Schinzel AC, Cho J, Hammerman PS, Verhaak RG, Hahn WC, Rajagopal J, Jacks T, Meyerson M. Integrated cistromic and expression analysis of amplified NKX2-1 in lung adenocarcinoma identifies LMO2 as a functional transcription target. Genes Dev. 2013; 27:197-210. [PMC free article: PMC3566312] [PubMed: 23322301]
- Willemsen MA, Breedveld GJ, Wouda S, Otten BJ, Yntema JL, Lammens M, de Vries BB. Brain-thyroid-lung syndrome: a patient with a severe multi-system disorder due to a de novo mutation in the thyroid transcription factor 1 gene. Eur J Pediatr, 2005;164:28-30. [PubMed: 15517377]
- Young LR, Deutsch GH, Bokulic R, Brody A, Nogee LM. A mutation in TTF1/NKX2.1 is associated with familial neuroendocrine cell hyperplasia of infancy (NEHI). Chest. 2013;144:1199-206. [PMC free article: PMC3787915] [PubMed: 23787483]
Publication Details
Author Information and Affiliations
Division of Movement Disorders
Department of Neurological Sciences
RUSH University Medical Center
Chicago, Illinois
Distinguished Chair in Movement Disorders
Director, Parkinson's Disease Center and Movement Disorders Clinic
Department of Neurology
Baylor College of Medicine
Houston, Texas
Publication History
Initial Posting: February 20, 2014; Last Update: June 29, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Patel NJ, Jankovic J. NKX2-1-Related Disorders. 2014 Feb 20 [Updated 2023 Jun 29]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.