Summary
Clinical characteristics.
Beta-propeller protein-associated neurodegeneration (BPAN) is typically characterized by early-onset seizures, infantile-onset developmental delay, intellectual disability, absent-to-limited expressive language, motor dysfunction (ataxia), and abnormal behaviors often similar to autism spectrum disorder. Seizure types including generalized (absence, tonic, atonic, tonic-clonic and myoclonic), focal with impaired consciousness, and epileptic spasms, as well as epileptic syndromes (West syndrome and Lennox-Gastaut syndrome) can be seen. With age seizures tend to resolve or become less prominent, whereas cognitive decline and movement disorders (progressive parkinsonism and dystonia) emerge as characteristic findings.
Diagnosis/testing.
The diagnosis of BPAN is established by identifying on molecular genetic testing:
- In females. A heterozygous WDR45 germline pathogenic variant;
- In males. Either a hemizygous WDR45 pathogenic variant or partial deletion of WDR45.
Somatic mosaicism has been reported in rare females (and possibly in 1 male).
Management.
Treatment of manifestations: Seizure management, tailored to the individual, may include anti-seizure (ASM), ketogenic diet, and/or vagus nerve stimulation. Developmental delays and intellectual disability are managed in infancy and early childhood with early intervention programs and in school-age children with individual education programs. Consultation with a developmental pediatrician to ensure appropriate services is recommended. Discussion about transition plans including financial, vocation/employment, and medical arrangements should begin at age 12 years. Consider alternative means of communication, such as an Augmentative and Alternative Communication (AAC) program, for individuals who have expressive language difficulties. Motor dysfunction in childhood is managed with physical therapy to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation). Occupational therapy may aid in writing, feeding, grooming, and dressing. For those with feeding difficulties, feeding therapy may be considered. For individuals with social/behavioral difficulties, consider applied behavior analysis (ABA) therapy, medical management strategies in conjunction with a developmental specialist, and pediatric psychiatry consultation to address aggressive or destructive behaviors. In adolescents/adults with parkinsonism, dystonia, and spasticity, the usual pharmacologic agents can be considered with the caveat that these therapies could exacerbate the cognitive deficits that also occur in this age group. Children with abnormal behaviors may qualify for and benefit from interventions used in treatment of autism spectrum disorder.
Surveillance: Routine follow up by a neurologist for medication management and interval assessment of ambulation, seizure activity, speech, and evidence of swallowing dysfunction. For those receiving dopaminergic drugs (for parkinsonism), monitor for adverse neuropsychiatric effects and disabling motor fluctuations and dyskinesias.
Pregnancy management: Use of ASM during pregnancy for women with a seizure disorder is generally recommended, despite an increased risk for adverse fetal outcome associated with some ASMs. Discussion of the risks and benefits of using a given anti-seizure medication during pregnancy ideally should take place prior to conception. Transitioning to a lower-risk medication may be possible.
Genetic counseling.
BPAN is inherited in an X-linked manner; to date, most affected individuals have been female, and the vast majority are simplex cases (i.e., a single occurrence in a family) resulting from a de novo WDR45 pathogenic variant. The exceptions include two familial cases, one with presumed maternal germline mosaicism and one with transmission of a WDR45 pathogenic variant by a phenotypically normal mother who demonstrated significantly skewed X-chromosome inactivation. Females with the typical BPAN phenotype do not reproduce. In rare instances, a female who is mildly affected may reproduce. Affected males do not reproduce. Once the WDR45 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for BPAN are possible.
Diagnosis
No formal diagnostic criteria for beta-propeller protein-associated neurodegeneration (BPAN) have been published.
Suggestive Findings
BPAN should be suspected in an individual with the following clinical and MRI findings.
Clinical Findings
Seizures characterized by the following:
- Features
- Onset in early childhood
- Development of multiple seizure types
- Seizures worse in early childhood, lessening with age
- Types
- Generalized (absence, tonic, atonic, tonic-clonic and myoclonic)
- Focal seizures with impaired consciousness
- Epileptic spasms
- Syndromes
- West syndrome
- Lennox-Gastaut syndrome
- Early-onset epilepsy with intellectual disability
Developmental delay / intellectual disability (with minimal or absent expressive language) that is relatively stable in childhood, followed by progressive loss of cognitive function beginning in adolescence or early adulthood
Progressive parkinsonism and dystonia, beginning in adolescence or early adulthood
Abnormal behaviors that overlap with Rett syndrome, including stereotypic hand movements, bruxism when awake, and abnormal sleep patterns
MRI Findings
Nonspecific hypomyelination and thin corpus callosum are seen in early childhood [Rathore et al 2014; Khalifa & Naffaa 2015; P Hogarth, unpublished data] and evolve to highly specific signal abnormalities in later childhood, adolescence, or early adulthood:
- T2-weighted or iron-sensitive sequences reveal hypointense signal in the substantia nigra and globus pallidus (Figure 1). Note: Although less prominent than in older individuals, this finding is documented in children as young as age five years [Hayflick et al 2013; Ichinose et al 2014; P Hogarth, unpublished data].
- T1-weighted sequences show a hyperintense "halo" surrounding a central linear region of signal hypointensity within the substantia nigra and cerebral peduncles (Figure 2).

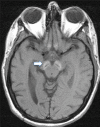
Figure 2.
T1 imaging demonstrates the hyperintense halo surrounding a central linear band of hypointensity in the substantia nigra and cerebral peduncles.
Cerebellar or cerebral atrophy may also be present at any age.
Establishing the Diagnosis
The diagnosis of BPAN is usually established in a female proband by identification of a heterozygous pathogenic (or likely pathogenic) variant in WDR45 by molecular genetic testing (Table 1). Note: (1) Somatic mosaicism has been documented in leukocyte DNA of healthy mothers of affected male sibs [Dufke et al 2014] and affected male/female sibs [Zarate et al 2016] (see Genetic Counseling and Molecular Genetics). (2) Per ACMG variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants.
The diagnosis of BPAN is established in a male proband by identification of a hemizygous pathogenic variant in WDR45 by molecular genetic testing (Table 1). Of note:
- A de novo 19.9-kb deletion that includes part of WDR45, all of an adjacent gene, and part of a third gene has been reported in a severely affected male [Abidi et al 2016] (see Molecular Genetics).
- In three families with recurrence of BPAN in sibs, affected males had inherited a germline pathogenic variant [Dufke et al 2014, Nakashima et al 2016, Zarate et al 2016] (see Molecular Genetics).
- Apparent somatic mosaicism for a WDR45 pathogenic variant in a male has been described [Haack et al 2012] (see Genetic Counseling and Molecular Genetics).
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel or single-gene testing) and genomic testing (comprehensive genome sequencing) depending on the phenotype.
Gene-targeted testing requires the clinician to determine which gene(s) are likely involved, whereas genomic testing may not. In some cases, the phenotype of BPAN may be distinctive enough to warrant single gene-targeted testing (see Option 1), whereas those with a phenotype that is indistinguishable from many other inherited disorders with intellectual disability and neurologic findings (including seizures) are more likely to be diagnosed using genomic testing (see Option 2).
Option 1. Gene-targeted testing. When the clinical and MRI findings suggest the diagnosis of BPAN, molecular genetic testing to consider includes the following:
- Single-gene testing. Sequence analysis of WDR45 is performed first. If no pathogenic variant is found, gene-targeted deletion/duplication analysis could be considered.
- A multigene panel that includes WDR45 and other genes of interest (see Differential Diagnosis). Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see Table 1). (5) If somatic mosaicism is suspected, discuss with the testing laboratory the ability of their methodology to detect mosaicism.
Option 2. Genomic testing. When the phenotype is indistinguishable from many other inherited disorders with intellectual disability and neurologic findings (including seizures), molecular genetic testing to consider includes the following:
- Comprehensive genomic testing (when available) including exome sequencing and genome sequencing. Note: These methods are generally suboptimal for detection of somatic mosaicism due to insufficient read depth of coverage.
- A multigene panel for inherited disorders with intellectual disability and neurologic findings
Table 1.
Molecular Genetic Testing Used in Beta Propeller Protein-Associated Neurodegeneration (BPAN)
Clinical Characteristics
Clinical Description
Beta-propeller protein-associated neurodegeneration (BPAN) typically presents with seizures, infantile-onset developmental delay, intellectual disability, absent-to-limited expressive language, and abnormal behaviors with Rett syndrome-like features. During adolescence and early adulthood (mean age 25 years; range 15-37 years) some of the hallmark childhood features (e.g., seizures) resolve or become less prominent, while cognitive decline and progressive parkinsonism and dystonia emerge as characteristic findings [Hayflick et al 2013, Nishioka et al 2015].
The majority of individuals with BPAN are female; however, some males have been reported. Significant phenotypic variability is observed in both males and females.
With increasing use of genomic testing, individuals with BPAN now tend to be identified at younger ages than those originally described by Haack et al [2012].
Core Clinical Features
Seizures are common during childhood, often starting as febrile seizures [Nishioka et al 2015]. Seizures are frequently multiform and include focal seizures with impaired consciousness, epileptic spasms [Hogarth, personal communication], and generalized seizures, including absence, atonic, myoclonic, and tonic-clonic [Hayflick et al 2013].
One young boy included in a cohort of children with early-onset epileptic encephalopathy had a de novo deletion of WDR45 and two contiguous genes (CCDC120 and PRAF2) and clinical findings consistent with BPAN (brain iron accumulation, dystonia, and spastic tetraparesis) [Abidi et al 2016].
Although seizures often require pharmacologic treatment and may be intractable, they often resolve or become less prominent with age.
Developmental delay / intellectual disability. Individuals with BPAN typically have developmental delay as children. The majority of children with BPAN have no expressive language; some develop limited language. Rarely, girls with BPAN can be high functioning with normal or nearly normal language development and less severe learning disabilities [Long et al 2015].
Onset of progressive dementia occurs between adolescence and early to middle adulthood (mean age 25 years; range 15-37 years) [Hayflick et al 2013]. Similar neurologic decline was observed in seven affected Japanese women between ages 29 to 39 years [Nishioka et al 2015].
In some instances, parents of children with BPAN have reported regression during early childhood; however, it is not clear whether this is due to the underlying disease, unrecognized seizures, or both [Hogarth, personal communication]. The aggressive epilepsy profile eventually seen in some children with BPAN may itself contribute to cognitive dysfunction, as in other epileptic encephalopathies.
Motor dysfunction. Children are typically clumsy and have a broad-based or ataxic gait. Some never learn to walk; others who achieve walking eventually become non-ambulatory. Some have mild spasticity that generally does not require treatment.
Fine motor skills are also impaired. Some also have limited purposeful hand use (reminiscent of Rett syndrome) that can contribute to functional impairments such as difficulty with dressing and use of utensils.
The neurologic deterioration that occurs during adolescence and early to middle adulthood also includes onset of movement disorders, dystonia, and parkinsonism [Hayflick et al 2013, Nishioka et al 2015].
Dystonia often starts in the upper extremities.
Parkinsonism is characterized by prominent bradykinesia, rigidity, freezing of gait, and postural instability. Tremor is not as common as in other forms of parkinsonism.
Abnormal behaviors that overlap with Rett syndrome include lack of purposeful hand movements, stereotypic hand movements such as repetitive midline hand-wringing, bruxism when awake, abnormal sleep patterns, features of autism spectrum disorder, and diminished response to pain [Haack et al 2012, Hayflick et al 2013, Ohba et al 2014, Khalifa & Naffaa 2015]. Some young girls have been noted to have episodes of deep breathing during waking hours [Authors, personal observation]. Several individuals with BPAN have been diagnosed with autism spectrum disorder – due in part to their limited language and social skills [Verhoeven et al 2014].
Other Features
Disordered sleep. Many families report that because their young children with BPAN have difficulty falling asleep and staying asleep, they sleep for only short periods of time. When performed, sleep studies revealed shortened mean sleep latency and abnormal REM sleep, hypersomnolence, hyposomnolence, and "dance-like" movements of the extremities with sleep onset [Hayflick et al 2013]. Unrecognized nocturnal seizure activity may contribute to abnormal sleep patterns.
Some adults with BPAN continue to have sleep difficulties or develop new manifestations such as waking and vocalizing during the night. Two of seven Japanese women had sleep problems as adults [Nishioka et al 2015].
Ophthalmologic findings including bilateral partial retinal colobomas, high myopia, astigmatism with myopia, spontaneous retinal detachment, and patchy loss of the pupillary ruff were observed in seven of 23 individuals [Hayflick et al 2013].
Bilateral optic atrophy has been described in one individual who also had bilateral sensorineural hearing loss [Rathore et al 2014].
Feeding and nutritional issues. Some infants and young children have feeding difficulties, most commonly texture sensitivity, oropharyngeal dysfunction, and aspiration. In some instances, GE reflux in young children has also required medical management. The neurologic deterioration that occurs in adolescence to early or middle adulthood frequently involves progressive feeding difficulty related to cognitive decline, dystonia, and parkinsonism and often results in significant weight loss.
Neuropathologic features. Findings in a woman who died from pneumonia at age 27 years included mild cerebellar atrophy, thinned cerebral peduncles, and dark gray-brown appearance of the substantia nigra and (to a lesser extent) the globus pallidus. The substantia nigra and globus pallidus stained strongly for iron and demonstrated numerous axonal spheroids – both findings consistent with the pathology of other types of neurodegeneration with brain iron accumulation. Numerous tau-positive neurofibrillary tangles were seen in several regions; no beta-amyloid plaques or Lewy bodies were observed [Hayflick et al 2013].
In a second, more advanced case the findings were similar, but with more extensive neuronal loss and tau pathology [Paudel et al 2015]. Recent, unpublished findings by the authors suggest a more complex pathology.
BPAN in Affected Males and Females
Although WDR45 is X-linked and in females is subject to X-chromosome inactivation, the clinical features of BPAN follow a pattern that is somewhat atypical for an X-linked disorder: while there are far fewer affected males than females, the phenotype is similar in the two sexes.
In the original report of 20 individuals, the three affected males had pathogenic variants predicted to render the protein nonfunctional (all frameshifts leading to premature stop codons). One of these males had evidence suggestive of somatic mosaicism [Haack et al 2012]. A hypothesis of somatic mosaicism of a WDR45 pathogenic variant can explain both the viability of these males and the similarity of their phenotype to that of affected heterozygous females who are functionally mosaic due to X-chromosome inactivation [Haack et al 2012, Saitsu et al 2013].
Males with germline pathogenic variants in WDR45 were originally predicted to be non-viable, but five such males from three families have since been reported [Dufke et al 2014, Nakashima et al 2016, Zarate et al 2016]. Two families had missense variants while the other had a 3-bp in-frame deletion.
The overall paucity of affected males relative to females suggests that germline pathogenic variants are rare in males and that affected male conceptuses are less likely to survive than female conceptuses. In two sets of male-female sibs with inherited WDR45 pathogenic variants, the males were more severely affected than the females [Nakashima et al 2016, Zarate et al 2016]; of note, one female showed strongly skewed X-chromosome inactivation with the abnormal allele being preferentially active [Zarate et al 2016].
In summary, the following factors are all proposed to contribute to the variability in phenotype and to the predominance of affected females relative to males:
- Somatic mosaicism in both sexes
- Skewed X-chromosome inactivation in females
- The specific type of pathogenic variant
Genotype-Phenotype Correlations
Germline nonsense variants are predicted to lead to non-viable males. The vast majority of pathogenic variants are de novo and private.
Nomenclature
The BPAN phenotype was recognized in the initial (2002) version of Pantothenate Kinase-Associated Neurodegeneration and in Gregory et al [2009]; however, naming it in a manner consistent with other forms of neurodegeneration with brain iron accumulation (NBIA) had to await discovery of the associated gene.
Although referenced in the literature, the term SENDA (static encephalopathy with neurodegeneration in adulthood) is no longer favored [Gregory & Hayflick 2011, Kruer et al 2012].
Prevalence
Similar to other forms of neurodegeneration with brain iron accumulation (NBIA), BPAN is an ultra-rare disorder. Approximately 45 affected individuals have been reported in the literature to date.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with pathogenic variants in WDR45.
Differential Diagnosis
In childhood, the core clinical features of BPAN – seizures, developmental delay / intellectual disability, movement disorders, and behavioral abnormalities – are highly variable between individuals and lack specificity, overlapping with many other genetic and sporadic (non-genetic) disorders. Early in the disease course, imaging abnormalities are similarly nonspecific, particularly if iron-sensitive sequences are not employed. The differential diagnosis at this stage is therefore broad, and includes Rett syndrome, Angelman syndrome, alpha-fucosidosis, and the epileptic encephalopathies of infancy and childhood:
- Dravet syndrome (See SCN1A Seizure Disorders.)
- West syndrome
- Lennox-Gastaut syndrome
- Epilepsy with electrical status during slow-wave sleep
- Gelastic epilepsy
- Kozhevnikov-Rasmussen syndrome
- Landau-Kleffner syndrome
- Early-infantile epileptic encephalopathy (See OMIM Developmental and Epileptic Encephalopathy Phenotypic Series to view genes associated with this phenotype in OMIM.)Note: Like BPAN, the epileptic encephalopathies present with multiple seizure types in the first months or years of life and are associated with severe neurocognitive deficits. Note that specific eponyms are applied to a particular clinical and electrographic profile and do not imply a specific etiology: many genetic and non-genetic etiologies can result in a given epilepsy syndrome.
If imaging reveals basal ganglia hypointensity on iron-sensitive sequences, the differential diagnosis narrows to the NBIA disorders and imaging phenocopies. These disorders can generally be distinguished from BPAN and each other by specific clinical and imaging features. See Neurodegeneration with Brain Iron Accumulation Disorders Overview.
Clinical features and the presence of high brain iron on MRI distinguish BPAN from other forms of childhood- or early adult-onset dystonia-parkinsonism, such as PINK1-related Parkinson disease and parkin-type Parkinson disease.
Neuropathologic findings (see Clinical Description, Other Features) can help distinguish BPAN from other forms of NBIA.
- In BPAN the pathologic changes found in the substantia nigra dominate those found in the globus pallidus (consistent with MRI findings), in contrast to other forms of NBIA in which the globus pallidus is more involved.
- BPAN can be distinguished by the lack of α-synuclein pathology that is abundant in MPAN and PLAN.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with beta-propeller protein-associated neurodegeneration (BPAN), the following evaluations are recommended:
- Complete neurologic examination and developmental assessment including evaluation of motor function, speech/language, swallowing, and sleep
- EEG monitoring, including sleep EEG, for any child with features (by history or observation) to suggest overt or occult seizures. Early referral to a neurology epilepsy sub-specialist is recommended if hypsarrhythmia or another aggressive electrographic pattern is found on EEG.
- Assessments by specialists in physical, occupational, and/or speech therapy
- Ophthalmologic examination
- Assessment of nutrition by a nutritionist or dietician
- Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Seizures. As seizure severity and complexity varies widely, management should be tailored to the individual.
Some children with BPAN may have no evidence of seizures or experience only rare febrile events and require no ongoing pharmacologic intervention. Others have severe, refractory epilepsy with multiple seizure types warranting the involvement of specialists in pediatric epilepsy and consideration of multiple therapeutic modalities, including anti-seizure medication, ketogenic diet, and/or vagus nerve stimulation.
Short-term treatment with ACTH, prednisolone, or vigabatrin should be considered early in the care of a child with epileptic spasms, based on the guidelines in the US consensus report from the International League Against Epilepsy [Pellock et al 2010].
Education of parents regarding common seizure presentations is appropriate. For information on non-medical interventions and coping strategies for parents or caregivers of children diagnosed with epilepsy, see Epilepsy Foundation Toolbox.
Developmental Delay / Intellectual Disability
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy. In the US, early intervention is a nationwide federally funded program available in all states.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. An evaluation will occur before placement to determine needed services and therapies and will be subsequently written into an individualized education plan (IEP).
Ages 5-21 years. In the US, an IEP should be developed by the local public school district based on each individuals' level of function. Affected children are permitted to remain in the public school district until age 21.
All ages. In the US, individuals are protected under IDEA (Individuals with Disabilities Education Act) and should receive free and appropriate public education in the least restricted environment.
Consultation with a developmental pediatrician is recommended to ensure that appropriate community, state, and educational agencies are involved and to support parents in maximizing quality of life. Developmental pediatricians can also provide assistance with transition to adulthood.
Consideration of private supportive therapies based on the affected individual's needs is recommended. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
In the US:
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction. Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
Consider use of durable medical equipment as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers)
For muscle tone abnormalities including hypertonia or dystonia, consider involving appropriate specialists to aid in management of baclofen, Botox® anti-parkinsonian medications, or orthopedic procedures.
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function (e.g., writing, feeding, grooming, and dressing).
Oral motor dysfunction. Assuming that the individual is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended for affected individuals who have difficulty feeding as a result of poor oral motor control.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC] for individuals who have expressive language difficulties.
Parkinsonism. Involvement of a neurologist specializing in movement disorders may be helpful once parkinsonism emerges in adolescents/adults.
In the treatment of parkinsonism, dystonia, and spasticity the usual pharmacologic agents can be considered with the caveat that these therapies could exacerbate the cognitive deficits that also occur in this age group.
Pharmacologic treatment of parkinsonism with dopaminergic medications usually has an initial striking benefit in males and females with improved motor function, affect, appetite, and interest in activities, followed by equally consistent disabling motor fluctuations ("on-off" phenomena) and dyskinesias [Hayflick et al 2013, Hogarth 2015].
As cognitive deficits progress in those with advanced disease, carbidopa/levodopa formulations may carry a lower risk of adverse neuropsychiatric effects than dopamine agonists. Amantadine may be helpful for dyskinesias, but also carries a risk (especially in higher doses) of adverse psychiatric effects such as hallucinations and agitation.
Pharmacologic treatment of dystonia and spasticity can include (if indicated) clonazepam or other benzodiazepines, oral baclofen, intramuscular botulinum toxin, and a trial of intrathecal baclofen. Trihexyphenydyl may also be useful for dystonia but may worsen cognitive dysfunction in advanced stages of the disease.
Social/Behavioral Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and is typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Other Features
Disordered sleep. Pharmacologic treatment of sleep abnormalities with melatonin, chloral hydrate, or benzodiazepines may be indicated.
Ophthalmologic findings. Eye findings are treated in a routine manner.
Other
- Use of a liquid nutritional supplement to help maintain weight as needed
- A gastric feeding tube to help maintain adequate weight (which becomes more difficult with disease progression) and to reduce the risk of aspiration pneumonia, if indicated
- Monitoring for complications of gastroesophageal reflux disease with institution of pharmacologic management if indicated. There are no data on the efficacy of fundoplication in BPAN.
- Over-the-counter fiber supplements and/or stool softeners to treat constipation, which is likely caused by a combination of immobility, diet, and medications
- Regular pulmonary hygiene to manage risk for pulmonary complications; tracheostomy as indicated when secretions are difficult to manage
Surveillance
The following are appropriate:
- Routine follow up by a neurologist for medication management and interval assessment of ambulation, seizure activity, speech, and swallowing. Often done every three to six months, follow up may be annual for those who are more stable.
- For those receiving dopaminergic drugs (for parkinsonism), monitoring for adverse neuropsychiatric effects and disabling motor fluctuations and dyskinesias
- Follow-up ophthalmologic examination every one to two years
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Because women with mild manifestations of BPAN could potentially reproduce, the potential for teratogenic effects of medication taken during pregnancy needs to be considered.
Therapies Under Investigation
Because BPAN falls in the category of NBIA and because individuals with BPAN have high brain iron, the possibility of chelation therapy is sometimes raised. The chelator deferiprone is currently under investigation for treatment of another NBIA disorder, PKAN. However, to date there is no evidence that iron chelation initiated early in the disease course prevents or delays the parkinsonian degeneration in adulthood or otherwise alters the course of PKAN. Nonetheless, results of these studies may inform its use in BPAN and/or lead to additional clinical trials.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Beta-propeller protein-associated neurodegeneration (BPAN) is inherited in an X-linked manner. To date, most individuals with BPAN have been female. Data suggest that one male with BPAN had apparent somatic mosaicism for a WDR45 pathogenic variant [Haack et al 2012].
Risk to Family Members
Parents of a proband. To date, the vast majority of reported individuals with BPAN have been simplex cases (i.e., a single occurrence in a family) and have had a de novo WDR45 pathogenic variant.
Recommended parental molecular genetic testing when a proband has an identified WDR45 pathogenic variant:
- Female proband. Molecular genetic testing of both parents
- Male proband. Molecular genetic testing of the mother only; the father of a male proband will neither have the disorder nor be hemizygous for the WDR45 pathogenic variant.
If a woman has more than one affected child and no other affected relatives, she most likely has germline (and possibly somatic) mosaicism.
- Presumed maternal germline mosaicism has been reported in a family with two affected sibs [Zarate et al 2016].
- In a second family, a phenotypically normal mother had the same WDR45 pathogenic variant as her sons with significant X-chromosome inactivation skewed towards the normal allele in her leukocyte DNA [Dufke et al 2014].
Sibs of a proband. To date, nearly all affected individuals have had a de novo WDR45 pathogenic variant, suggesting a low risk to sibs.
- Female proband. The risk to sibs depends on the genetic status of the parents.
- Male proband. The risk to sibs depends on the genetic status of the mother.
If a proband represents a simplex case (i.e., a single occurrence in a family) and if the WDR45 pathogenic variant cannot be detected in the parent's leukocyte DNA (female proband) or in the mother's leukocyte DNA (male proband), the risk to sibs is presumed to be slightly greater than that of the general population (though still <1%) because of the possibility of germline mosaicism [Zarate et al 2016].
Offspring of a proband
- Female proband. Females with the typical BPAN phenotype do not reproduce. In rare cases, a female who is mildly affected may reproduce.
- One very mildly affected female with somatic and, presumably, germline mosaicism has reproduced [Zarate et al 2016]. The likelihood that a mildly affected female with somatic and germline mosaicism would transmit a pathogenic variant to offspring may be less than 50% [Haack et al 2012, Zarate et al 2016]. However, the potential reduction in recurrence risk is not quantifiable. Male fetuses are less likely to survive than female fetuses.
- One very mildly affected female with skewed X-chromosome inactivation in leukocyte DNA favoring the normal WDR45 allele has reproduced [Dufke et al 2014]. In this instance, the chance to transmit the pathogenic variant to offspring is 50%. A male conceptus may be less likely to survive.
- Male proband. Affected males do not reproduce.
Other family members. Given that most probands with BPAN reported to date have the disorder as a result of a de novo WDR45 pathogenic variant, the risk to other family members is presumed to be low.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of genetic status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to parents of affected individuals.
Prenatal Testing and Preimplantation Genetic Testing
Once the WDR45 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for BPAN are possible.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- NBIA AllianceEmail: Info@NBIAalliance.org
- NBIA Disorders Association
- NBIAcureCenter of Excellence for NBIA Clinical Care and ResearchInternational Registry for NBIA and Related DisordersOregon Health & Science UniversityEmail: info@nbiacure.org
- Treat Iron-Related Childhood Onset Neurodegeneration (TIRCON)GermanyEmail: TIRCON@med.uni-muenchen.de
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Beta-Propeller Protein-Associated Neurodegeneration: Genes and Databases
Table B.
OMIM Entries for Beta-Propeller Protein-Associated Neurodegeneration (View All in OMIM)
Gene structure. WDR45 contains 12 exons; the first two are non-coding. The longest transcript variant is NM_007075.3; a variant transcript NM_001029896.1 has an alternate 5'UTR and lacks an in-frame three-nucleotide segment in the coding region, thus encoding a protein isoform with one less internal amino acid. See Table A, Gene for details on transcript variants.
Pathogenic variants. Pathogenic variants have been documented in coding exons 3 to 12. All categories of variants have been reported including missense, nonsense, splicing, and intragenic deletion/insertion (i.e., indel) (Table A, HGMD). Of note, a 3-bp in-frame deletion has been reported in male/female sibs and their mother, who was mosaic for this variant [Zarate et al 2016]. Apparent mosaicism was also found in an affected male [Haack et al 2012].
The majority of reported variants are predicted to be loss-of-function variants [Haack et al 2013]; the pathogenic basis for missense variants and the 3-bp in-frame deletion are unclear.
Additionally, a 19.9-kb de novo, non-mosaic deletion of WDR45 exons 3-12 as well as two adjacent genes CCDC120 and PRAF2 (partial and complete deletion, respectively) was reported in an affected male [Abidi et al 2016].
Nearly all pathogenic variants are de novo; most are unique and appear to occur across ethnic groups at the same frequency.
Normal gene product. WDR45 encodes WD repeat domain phosphoinositide-interacting protein 4 (WIPI4), which has WD40 repeat units. WIPI4 is a member of a large family of proteins with repeating units containing a conserved core of 40+ amino acids that terminate with tryptophan-aspartate (W-D) residues [Li & Roberts 2001]. These proteins assume a highly symmetric, seven-bladed, beta-propeller platform structure; hence the name of the disorder, beta-propeller protein-associated neurodegeneration (BPAN). This structure supports specific protein-protein interactions and has a putative role in autophagy. Specifically WIPI4 is implicated in binding – or associating – with the known autophagy proteins ATG2A and ATG2B [Proikas-Cezanne et al 2004, Behrends et al 2010]; however, the exact role of WDR45 in the autophagic process remains to be elucidated.
Abnormal gene product. Lymphoblasts and brain tissue from individuals with WDR45 pathogenic variants demonstrate evidence of dysregulation of autophagic flux [Saitsu et al 2013, Paudel et al 2015]. A growing body of literature suggests dysregulation of autophagy as a mechanism in the pathophysiology of neurodegeneration [Nassif & Hetz 2012]. In normal neurons, basal autophagy helps maintain protein quality control and organellar homeostasis. It has been proposed that injured or stressed cells rely on autophagy to clear damaged cell components and other by-products, and that neurons are particularly vulnerable to defects in autophagy because of the large expanse of dendritic and axonal cytoplasm that must be traversed in order to reach lysosomes concentrated near the cell body [Nixon 2013].
Chapter Notes
Author Notes
The NBIAcure website, developed and maintained by Drs Susan Hayflick and Penny Hogarth and their team, contains up-to-date information about BPAN and the other NBIA disorders, geared for lay persons, clinicians, and researchers. Information about current research and opportunities to enroll online are available here.
Acknowledgments
Research reported in this publication was supported by a grant awarded to Dr Hogarth by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number UL1TR000128. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Revision History
- 16 February 2017 (bp) Review posted live
- 9 February 2016 (ag) Original submission
References
Literature Cited
- Abidi A, Mignon-Ravix C, Cacciagli P, Girard N, Milh M, Villard L. Early-onset epileptic encephalopathy as the initial clinical presentation of WDR45 deletion in a male patient. Eur J Hum Genet. 2016;24:615–8. [PMC free article: PMC4929878] [PubMed: 26173968]
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. [PMC free article: PMC2901998] [PubMed: 20562859]
- Dufke A, Grasshoff U, Dufke C, Kalscheuer V, Schroeder C, Beck-Wodl S, Tzschach A, Nagele T, Riess O, Bauer P, Krageloh-Mann I. NGS based whole X-exome analysis reveals a familial WDR45 missense mutation in 3 males with intellectual disability and brain iron accumulation. Abstract P08.53-S. Milan, Italy: European Society of Human Genetics Conference. 2014. Abstract available online.
- Gregory A, Hayflick SJ. Genetics of neurodegeneration with brain iron accumulation. Curr Neurol Neurosci Rep. 2011;11:254–61. [PMC free article: PMC5908240] [PubMed: 21286947]
- Gregory A, Polster BJ, Hayflick SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet. 2009;46:73–80. [PMC free article: PMC2675558] [PubMed: 18981035]
- Haack TB, Hogarth P, Kruer MC, Gregory A, Wieland T, Schwarzmayr T, Graf E, Sanford L, Meyer E, Kara E, Cuno SM, Harik SI, Dandu VH, Nardocci N, Zorzi G, Dunaway T, Tarnopolsky M, Skinner S, Frucht S, Hanspal E, Schrander-Stumpel C, Héron D, Mignot C, Garavaglia B, Bhatia K, Hardy J, Strom TM, Boddaert N, Houlden HH, Kurian MA, Meitinger T, Prokisch H, Hayflick SJ. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet. 2012;91:1144–9. [PMC free article: PMC3516593] [PubMed: 23176820]
- Haack TB, Hogarth P, Gregory A, Prokisch H, Hayflick SJ. BPAN: the only X-linked dominant NBIA disorder. Int Rev Neurobiol. 2013;110:85–90. [PubMed: 24209435]
- Hayflick SJ, Kruer MC, Gregory A, Haack TB, Kurian MA, Houlden HH, Anderson J, Boddaert N, Sanford L, Harik SI, Dandu VH, Nardocci N, Zorzi G, Dunaway T, Tarnopolsky M, Skinner S, Holden KR, Frucht S, Hanspal E, Schrander-Stumpel C, Mignot C, Héron D, Saunders DE, Kaminska M, Lin JP, Lascelles K, Cuno SM, Meyer E, Garavaglia B, Bhatia K, de Silva R, Crisp S, Lunt P, Carey M, Hardy J, Meitinger T, Prokisch H, Hogarth P. β-propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain. 2013;136:1708–17. [PMC free article: PMC3673459] [PubMed: 23687123]
- Hogarth P. Neurodegeneration with brain iron accumulation: diagnosis and management. J Mov Disord. 2015;8:1–13. [PMC free article: PMC4298713] [PubMed: 25614780]
- Ichinose Y, Miwa M, Onohara A, Obi K, Shindo K, Saitsu H, Matsumoto N, Takiyama Y. Characteristic MRI findings in beta-propeller protein-associated neurodegeneration (BPAN). Neurol Clin Pract. 2014;4:175–7. [PMC free article: PMC4001181] [PubMed: 24790802]
- Khalifa M, Naffaa L. Exome sequencing reveals a novel WDR45 frameshift mutation and inherited POLR3A heterozygous variants in a female with a complex phenotype and mixed brain MRI findings. Eur J Med Genet. 2015;58:381–6. [PubMed: 26096995]
- Kruer MC, Boddaert N, Schneider SA, Houlden H, Bhatia KP, Gregory A, Anderson JC, Rooney WD, Hogarth P, Hayflick SJ. Neuroimaging features of neurodegeneration with brain iron accumulation. AJNR Am J Neuroradiol. 2012;33:407–14. [PMC free article: PMC7966445] [PubMed: 21920862]
- Li D, Roberts R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell Mol Life Sci. 2001;58:2085–97. [PMC free article: PMC11337334] [PubMed: 11814058]
- Long M, Abdeen N, Geraghty MT, Hogarth P, Hayflick S, Venkateswaran S. Novel WDR45 mutation and pathognomonic BPAN imaging in a young female with mild cognitive delay. Pediatrics. 2015;136:e714–7. [PubMed: 26240209]
- Nakashima M, Takano K, Tsuyusaki Y, Yoshitomi S, Shimono M, Aoki Y, Kato M, Aida N, Mizuguchi T, Miyatake S, Miyake N, Osaka H, Saitsu H, Matsumoto N. WDR45 mutations in three male patients with West syndrome. J Hum Genet. 2016;61:653–61. [PubMed: 27030146]
- Nassif M, Hetz C. Autophagy impairment: a crossroad between neurodegeneration and tauopathies. BMC Biol. 2012;10:78. [PMC free article: PMC3448508] [PubMed: 22999309]
- Nishioka K, Oyama G, Yoshino H, Li Y, Matsushima T, Takeuchi C, Mochizuki Y, Mori-Yoshimura M, Murata M, Yamasita C, Nakamura N, Konishi Y, Ohi K, Ichikawa K, Terada T, Obi T, Funayama M, Saiki S, Hattori N. High frequency of beta-propeller protein-associated neurodegeneration (BPAN) among patients with intellectual disability and young-onset parkinsonism. Neurobiol Aging. 2015;36:2004.e9–15. [PubMed: 25744623]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–97. [PubMed: 23921753]
- Ohba C, Nabatame S, Iijima Y, Nishiyama K, Tsurusaki Y, Nakashima M, Miyake N, Tanaka F, Ozono K, Saitsu H, Matsumoto N. De novo WDR45 mutation in a patient showing clinically Rett syndrome with childhood iron deposition in brain. J Hum Genet. 2014;59:292–5. [PubMed: 24621584]
- Paudel R, Li A, Wiethoff S, Bandopadhyay R, Bhatia K, de Silva R, Houlden H, Holton JL. Neuropathology of Beta-propeller protein associated neurodegeneration (BPAN): a new tauopathy. Acta Neuropathol Commun. 2015;3:39. [PMC free article: PMC4486689] [PubMed: 26123052]
- Pellock JM, Hrachovy R, Shinnar S, Baram TZ, Bettis D, Dlugos DJ, Gaillard WD, Gibson PA, Holmes GL, Nordl DR, O'Dell C, Shields WD, Trevathan E, Wheless JW. Infantile spasms: a U.S. consensus report. Epilepsia. 2010;51:2175–89. [PubMed: 20608959]
- Pont-Kingdon G, Gedge F, Wooderchak-Donahue W, Schrijver I, Weck KE, Kant JA, Oglesbee D, Bayrak-Toydemir P, Lyon E, et al. Design and analytical validation of clinical DNA sequencing assays. Arch Pathol Lab Med. 2012;136:41–6. [PubMed: 22208486]
- Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A, Nordheim A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene. 2004;23:9314–25. [PubMed: 15602573]
- Rathore GS, Schaaf CP, Stocco AJ. Novel mutation of the WDR45 gene causing beta-propeller protein-associated neurodegeneration. Mov Disord. 2014;29:574–5. [PubMed: 24610255]
- Saitsu H, Nishimura T, Muramatsu K, Kodera H, Kumada S, Sugai K, Kasai-Yoshida E, Sawaura N, Nishida H, Hoshino A, Ryujin F, Yoshioka S, Nishiyama K, Kondo Y, Tsurusaki Y, Nakashima M, Miyake N, Arakawa H, Kato M, Mizushima N, Matsumoto N. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet. 2013;45:445–9. [PubMed: 23435086]
- Verhoeven WM, Egger JI, Koolen DA, Yntema H, Olgiati S, Breedveld GJ, Bonifati V, van de Warrenburg BP. Beta-propeller protein-associated neurodegeneration (BPAN), a rare form of NBIA: novel mutations and neuropsychiatric phenotype in three adult patients. Parkinsonism Relat Disord. 2014;20:332–6. [PubMed: 24368176]
- Zarate YA, Jones JR, Jones MA, Millan F, Juusola J, Vertino-Bell A, Schaefer GB, Kruer MC. Lessons from a pair of siblings with BPAN. Eur J Hum Genet. 2016;24:1080–3. [PMC free article: PMC5070893] [PubMed: 26577041]
Publication Details
Author Information and Affiliations
Portland, Oregon
London, United Kingdom
Munich, Germany
Portland, Oregon
Portland, Oregon
Publication History
Initial Posting: February 16, 2017.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Gregory A, Kurian MA, Haack T, et al. Beta-Propeller Protein-Associated Neurodegeneration. 2017 Feb 16. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

