Summary
Clinical characteristics.
Fatty acid hydroxylase-associated neurodegeneration (FAHN) is characterized early in the disease course by central nervous system involvement including corticospinal tract involvement (spasticity), mixed movement disorder (ataxia/dystonia), and eye findings (optic atrophy, oculomotor abnormalities), and later in the disease course by progressive intellectual impairment and seizures. With disease progression, dystonia and spasticity compromise the ability to ambulate, leading to wheelchair dependence. Life expectancy is variable. FAHN is considered to be a subtype of neurodegeneration with brain iron accumulation (NBIA).
Diagnosis/testing.
The diagnosis of FAHN is established in a proband with suggestive findings and typically by identification of biallelic FA2H pathogenic variants on molecular genetic testing; however, on occasion uniparental disomy (UDP) is causative.
Management.
Treatment of manifestations: Symptomatic treatment focuses primarily on the dystonia, which can be debilitating. Therapies used with varying success include the oral medications baclofen, tizanidine, dantrolene, and anticholinergics; injection of botulinum toxin targeting abnormal co-contraction of selected muscle groups; and ablative pallidotomy or thalamotomy. Attention should be given to nutritional status and feeding.
Independence should be encouraged when possible through use of adaptive aids (e.g., walker or wheelchair for gait abnormalities, augmentative communication devices) and appropriate community resources (e.g., financial services, programs for the visually impaired, special education).
Surveillance: Regular assessment of nutritional status and swallowing, vision, mobility and environmental adaptations, speech, and communication needs.
Genetic counseling.
Genetic counseling for fatty acid hydroxylase-associated neurodegeneration (FAHN) depends on the causative genetic mechanism: FAHN caused by transmission of one pathogenic variant from each parent is inherited in an autosomal recessive manner; FAHN caused by transmission of two pathogenic variants from one parent (as the result of uniparental disomy [UPD] for chromosome 16) is a de novo event and is unlikely to recur.
Autosomal recessive inheritance: At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. If the pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
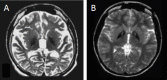
Fatty acid hydroxylase-associated neurodegeneration (FAHN) should be considered in individuals with the following clinical findings, neuroimaging findings, and family history (see Figure 1).
Clinical findings
- Onset within the first or second decade of life
- Corticospinal tract involvement:
- Spastic paraplegia or quadriplegia (commonly given a clinical diagnosis of hereditary spastic paraplegia)
- Pyramidal tract signs (hypereflexia, clonus, Babinski sign, Hoffmann sign)
- Movement disorder including one or both of the following:
- Dystonia
- Ataxia
- Dysarthria
- Dysphagia
- Eye findings:
- Optic atrophy manifest as progressive loss of visual acuity, sectoral visual field loss, and impaired color vision
- In some individuals: strabismus, lateral-beating nystagmus, and supranuclear gaze palsy
- Epilepsy
- Cognitive decline
Neuroimaging findings. Brain MRI findings typically may include (in order of likelihood):
- On T2-weighted images: variable unilateral or bilateral symmetric white matter hyperintensity that may affect both periventricular and subcortical white matter, and may become confluent with time. U-fibers and cerebellar white matter appear to be affected to a lesser degree.
- Progressive atrophy of the cerebellar hemispheres, vermis, pons, medulla and spinal cord
- Thinning of the corpus callosum
- Optic atrophy
- T2-weighted hypointensity of the globus pallidus (may display blooming on T2*-weighted images).Note: T2-weighted hypointensity coupled with an extrapyramidal movement disorder and intellectual decline is suggestive of a neurodegeneration with brain iron accumulation (NBIA) disorder (see Differential Diagnosis). The "eye of the tiger" sign, pathognomonic for pantothenate kinase-associated neurodegeneration (PKAN), another form of NBIA, is not seen in FAHN.
Family history is consistent with autosomal recessive inheritance, including parental consanguinity.
Establishing the Diagnosis
The diagnosis of FAHN is established in a proband with suggestive findings and biallelic FA2H pathogenic (or likely pathogenic) variants on molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic FA2H variants of uncertain significance (or of one known FA2H pathogenic variant and one FA2H variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with findings indistinguishable from other neurodegenerative disorders with pyramidal and extrapyramidal involvement are more likely to be diagnosed with comprehensive genomic testing (see Option 2).
Option 1
When the phenotypic and imaging findings suggest the diagnosis of FAHN or a similar type of neurodegeneration with brain iron accumulation (see NBIA Overview), molecular genetic testing is most often a multigene panel.
A multigene panel that includes FA2H and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the phenotype is indistinguishable from many other inherited neurodegenerative disorders with pyramidal and extrapyramidal involvement, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Fatty Acid Hydroxylase-Associated Neurodegeneration (FAHN)
Clinical Characteristics
Clinical Description
Fatty acid hydroxylase-associated neurodegeneration (FAHN) is characterized early in the disease course by central nervous system involvement including corticospinal tract involvement (spasticity), mixed movement disorder (ataxia/dystonia), eye findings (optic atrophy, oculomotor abnormalities), and later in the disease course by progressive intellectual impairment and seizures. FAHN is a subtype of neurodegeneration with brain iron accumulation (NBIA) but is also included under the classifications of leukodystrophies and hereditary spastic paraplegias.
Of note, leukodystrophy and hereditary spastic paraplegia 35 (HSP35), two phenotypes previously considered distinct disorders based on clinical findings [Dick et al 2008, Edvardson et al 2008], are now included in the phenotypic spectrum of FAHN based on molecular genetic findings [Dick et al 2010, Kruer et al 2010].
The most frequent presenting finding is a subtle change in gait that may lead to increasingly frequent falls. This typically occurs in childhood or adolescence and may be the result of focal dystonia and/or corticospinal tract involvement.
The degree of spasticity resulting from corticospinal tract involvement can vary among persons with FAHN. Individuals affected to a lesser degree may develop spastic paraparesis but retain the ability to walk, while individuals with more severe disease may demonstrate a spastic quadriplegic pattern of disability and lose their ability to ambulate, instead relying on a wheelchair.
Dystonia may begin focally (e.g., affecting one foot) but typically spreads to assume a generalized pattern. The degree of dystonia seen in FAHN is generally milder than that in other forms of NBIA, such as PKAN, in which status dystonicus occurs.
Ataxia typically begins in childhood or adolescence and may emerge along with dystonia and/or spasticity. The ataxia that occurs in FAHN may affect both axial and appendicular function and, along with both dystonia and spasticity, can markedly impair gait.
Dysarthria may be prominent in FAHN. In some individuals, expressive speech can be impaired to the point of anarthria. Dysphagia, potentially necessitating gastrostomy tube placement, can also occur.
Optic atrophy in FAHN may begin as a subtle loss of visual acuity in childhood, but may progress to the point of functional blindness. The oculomotor abnormalities seen in FAHN may impair functional vision as well.
A few individuals with FAHN and peripheral neuropathy have been reported [Donkervoort et al 2014, Zaki et al 2015].
Seizures are not typically seen in the early stages of disease, but may occur later in the disease course. When present, seizures (which tend to be complex partial or generalized) are typically infrequent and respond relatively well to anticonvulsants.
While progressive intellectual impairment occurs in most persons with FAHN, more information on the cognitive phenotype and natural history are needed. Serial assessments have documented cognitive decline in two individuals [Tonelli et al 2012]. One report suggests a psychiatric component to FAHN based on findings of anxiety, depression, and bipolar disorder in affected sibs [Magariello et al 2017].
Although the neurodegeneration in FAHN is progressive, declines may be intermittent and punctuated by periods of relative clinical stability. However, lost skills are not usually regained. With disease progression, dystonia and spasticity compromise the ability to ambulate, leading to wheelchair dependence.
Life expectancy. Although premature death often occurs in the 20s or 30s secondary to a combination of nutrition-related immunodeficiency and respiratory compromise, life expectancy is variable.
Neuropathologic features for FAHN have not yet been reported. Bone marrow biopsy, although not necessary for diagnosis, may demonstrate accumulation of granular histiocytes.
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been observed for pathogenic variants in FA2H.
Nomenclature
Historically, the FA2H-related phenotype has been classified as either a genetic leukodystrophy [Vanderver et al 2015] or a hereditary spastic paraplegia (SPG35) (see OMIM 612319) [Dick et al 2008, Soehn et al 2016].
The authors prefer to refer to the FA2H-related phenotype as fatty acid hydroxylase-associated neurodegeneration because this term refers to the genetic basis of the disorder and encompasses all clinical classifications.
Prevalence
No reliable prevalence data are available. However, the prevalence is estimated to be lower than one in 1,000,000.
Genetically Related (Allelic) Disorders
No other phenotypes are known to be associated with biallelic FA2H pathogenic variants.
Differential Diagnosis
Fatty acid hydroxylase-associated neurodegeneration (FAHN) is a neurodegenerative disorder that shows clinical overlap with other early-onset neurodegenerative disorders. Disorders that may exhibit clinical and neuroimaging features similar to those seen in FAHN are summarized in Table 2.
Table 2.
Disorders to Consider in the Differential Diagnosis of FAHN
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease in an individual diagnosed with fatty acid hydroxylase-associated neurodegeneration (FAHN), the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with Fatty Acid Hydroxylase-Associated Neurodegeneration (FAHN)
Treatment of Manifestations
Eyes
Refer those with visual impairment to appropriate community resources.
Feeding
Once swallowing evaluation and nutrition assessments indicate that the individual cannot maintain adequate nutrition and/or avoid the risk of aspiration with oral feeding, gastrostomy tube placement is indicated.
Neurologic
Pharmacologic and surgical interventions are focused on palliation of symptoms.
Dystonia and spasticity can be debilitating. Symptomatic treatments used with varying success include the following:
- Oral trihexyphenidyl, baclofen, tizanidine, benzodiazepines, and/or dantrolene. Of note, while levodopa could potentially provide benefit, it often does not; thus, a trial is reasonable, but should only be continued if there is clear benefit.
- Intramuscular botulinum toxin targeting abnormal co-contraction of selected muscle groups
- Ablative pallidotomy or thalamotomy. Dystonia may return despite this aggressive measure [Justesen et al 1999].
Ataxia. Therapies for cerebellar ataxia can include use of weighted gloves to assist with dysmetria. Note that riluzole, recently recommended for cerebellar ataxia [Ristori et al 2010], has not to the authors' knowledge undergone a therapeutic trial in FAHN.
Seizures. Seizures respond well to traditional management with anti-seizure medication.
Education of parents regarding common seizure presentations is appropriate. For information on non-medical interventions and coping strategies for parents or caregivers of children diagnosed with epilepsy, see Epilepsy Foundation Toolbox.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility.
- Consider use of durable medical equipment as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties.
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy. In the US, early intervention is a federally funded program available in all states.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed.
Ages 5-21 years
- In the US and Canada, an IEP based on the individual’s level of function should be developed by the local public school district. Affected children are permitted to remain in the public school district until age 21.
- Discussion about transition plans including financial and medical arrangements should begin at age 12 years. Developmental pediatricians can provide assistance with transition to adulthood.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies and to support parents in maximizing quality of life.
Consideration of private supportive therapies based on the affected individual's needs is recommended. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
In the US:
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Surveillance
The following should be performed on a regular basis:
- Swallowing evaluation, nutrition assessment, and monitoring of height and weight to screen for evidence of worsening nutritional status
- Ophthalmologic assessment with particular attention to visual acuity.
- Assessment of mobility, self-help skills, and activities of daily living and need for adaptive devices
- Assessment of speech and communication needs
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Iron chelation. Interest in iron chelation has reemerged as trials using deferiprone have been published in other disorders of brain iron accumulation, including Friedreich ataxia [Boddaert et al 2007] and superficial siderosis [Levy & Llinas 2011]. Deferiprone can cross the blood-brain barrier and remove intracellular iron. A multicenter, placebo controlled, double-blind trial comparing the efficacy and safety of 18 months of treatment with deferiprone versus placebo in patients with PKAN was completed in January of 2017 (see Clinical Trials). Data are currently being analyzed. Results, when published, may be generalizable to other NBIA disorders. However, long-term clinical trials of deferiprone in specific forms of NBIA will be necessary to further assess safety and efficacy.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Genetic counseling for fatty acid hydroxylase-associated neurodegeneration (FAHN) depends on the causative genetic mechanism:
- FAHN caused by transmission of one pathogenic variant from each parent is inherited in an autosomal recessive manner.
- FAHN caused by transmission of two pathogenic variants from one parent (as the result of uniparental disomy [UPD] for chromosome 16) is a de novo event and is unlikely to recur.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
- The parents of an affected individual are obligate heterozygotes (i.e., carriers of one FA2H pathogenic variant).
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. The offspring of an individual with FAHN are obligate heterozygotes (carriers) for a pathogenic variant in FA2H.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an FA2H pathogenic variant.
Carrier detection. Carrier testing for at-risk relatives requires prior identification of the FA2H pathogenic variants in the family.
Uniparental Disomy of Chromosome 16 (UPD16) – Risk to Family Members
Parents, sibs, and offspring of a proband
- The risk to parents, sibs, and offspring of an individual with FAHN caused by UPD16 is unlikely to be higher than the risk to the general population, as UPD16 is a de novo, typically non-recurrent event.
- If the proband has a chromosome abnormality in addition to UPD16, the risk to parents, sibs, and offspring is related to the specific abnormality identified in the proband.
- Note: UPD for chromosome 16 has led to four published cases of FAHN [Soehn et al 2016].
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Once the FA2H pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- NBIA AllianceEmail: Info@NBIAalliance.org
- NBIA CanadaPO Box 29South River Newfoundland and Labrador A0A 3W0CanadaPhone: 709-786-7248Email: info@NBIAalliance.org
- NBIA Disorders Association
- National Institute of Neurological Disorders and Stroke (NINDS)
- Spastic Paraplegia Foundation, Inc.Phone: 877-773-4483Email: information@sp-foundation.org
- NBIAcureCenter of Excellence for NBIA Clinical Care and ResearchInternational Registry for NBIA and Related DisordersOregon Health & Science UniversityEmail: info@nbiacure.org
- Treat Iron-Related Childhood Onset Neurodegeneration (TIRCON)GermanyEmail: TIRCON@med.uni-muenchen.de
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Fatty Acid Hydroxylase-Associated Neurodegeneration: Genes and Databases
Table B.
OMIM Entries for Fatty Acid Hydroxylase-Associated Neurodegeneration (View All in OMIM)
Molecular Pathogenesis
Human FA2H encodes the fatty acid 2-hydroxylase, an enzyme that alpha-hydroxylates nascent fatty acids that are subsequently incorporated into several diverse lipid species. Pathogenic variants in FA2H are believed to lead to fatty acid hydroxylase-associated neurodegeneration (FAHN) through a loss-of-function mechanism. This is supported by the identification of variants that predict premature truncation of the protein [Kruer et al 2010], as well as in vitro evidence of decreased α-hydroxylation activity [Dick et al 2010] and decreased protein abundance [Kruer et al 2010] secondary to pathogenic variants associated with clinical FAHN.
In the peripheral nervous system, there is evidence that a second fatty acid-hydroxylating enzyme (perhaps phytanoyl coA 2-hydroxylase) may compensate for loss of FA2H activity [Edvardson et al 2008]. However, in the central nervous system, this redundancy does not seem to exist. A decrease in 2-OH fatty acids may lead to abnormal white matter, as 2-OH sphingomyelin is an important myelin constituent [Eckhardt et al 2005]. Deficiency of 2-OH fatty acids may also lead to an abnormal increase in membrane fluidity [Guo et al 2010], with implications for the regulation of autophagy via the endosomal-lysosomal system. An important cell-cycle signaling role for FA2H has also been shown [Alderson & Hama 2009]. However, further investigation is needed to better characterize the mechanism(s) by which loss of functional FA2H may lead to FAHN.
Gene structure. The reference sequence NM_024306.4 has seven exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. Most sequence variants identified to date in persons with FAHN are unique variants. Based on data to date, partial or whole-gene deletions/duplications appear rare [Pierson et al 2012]. A study of uniparental disomy in nonconsanguineous families with HSP identified four unrelated individuals with FAHN. Uniparental disomy for chromosome 16 had led to homozygosity for FA2H pathogenic variants in these individuals [Soehn et al 2016].
Normal gene product. FA2H encodes a 43-kd protein of 372 amino acid residues that is a functional fatty acid hydroxylase [Alderson et al 2004]. This protein is localized to the endoplasmic reticulum and requires iron as a cofactor. Hydroxy fatty acids have important structural and signal transduction roles.
Abnormal gene product. Pathogenic FA2H missense variants have been shown to lead to decreased protein abundance or enzyme activity. FA2H frameshift variants and a multiexon deletion support the notion of decreased function or loss of function as a cause of FAHN.
Chapter Notes
Author History
Allison Gregory, MS (2011-present)
Susan J Hayflick, MD (2011-present)
Michael C Kruer, MD, University of South Dakota Sanford School of Medicine (2011-2018)
Sunita Venkateswaran, MD (2018-present)
Revision History
- 27 September 2018 (bp) Comprehensive update posted live
- 20 September 2012 (cd) Revision: deletion/duplication analysis available clinically
- 28 June 2011 (me) Review posted live
- 3 January 2011 (mk) Original submission
References
Literature Cited
- Alderson NL, Hama H. Fatty acid 2-hydroxylase regulates cAMP-induced cell cycle exit in D6P2T schwannoma cells. J Lipid Res. 2009;50:1203–8. [PMC free article: PMC2681402] [PubMed: 19171550]
- Alderson NL, Rembiesa BM, Walla MD, Bielawska A, Bielawski J, Hama H. The human FA2H gene encodes a fatty acid 2-hydroxylase. J Biol Chem. 2004;279:48562–8. [PubMed: 15337768]
- Boddaert N, Le Quan Sang KH, Rötig A, Leroy-Willig A, Gallet S, Brunelle F, Sidi D, Thalabard JC, Munnich A, Cabantchik ZI. Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood. 2007;110:401–8. [PubMed: 17379741]
- Dick KJ, Al-Mjeni R, Baskir W, Koul R, Simpson MA, Patton MA, Raeburn S, Crosby AH. A novel locus for an autosomal recessive hereditary spastic paraplegia (SPG35) maps to 16q21-q23. Neurology. 2008;71:248–52. [PubMed: 18463364]
- Dick KJ, Eckhardt M, Paisán-Ruiz C, Alshehhi AA, Proukakis C, Sibtain NA, Maier H, Sharifi R, Patton MA, Bashir W, Koul R, Raeburn S, Gieselmann V, Houlden H, Crosby AH. Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35). Hum Mutat. 2010;31:E1251–60. [PubMed: 20104589]
- Donkervoort S, Dastgir J, Hu Y, Zein WM, Marks H, Blackstone C, Bönnemann CG. Phenotypic variability of a likely FA2H founder mutation in a family with complicated hereditary spastic paraplegia. Clin Genet. 2014;85:393–5. [PMC free article: PMC5030767] [PubMed: 23745665]
- Eckhardt M, Yaghootfam A, Fewou SN, Zöller I, Gieselmann V. A mammalian fatty acid hydroxylase responsible for the formation of alpha-hydroxylated galactosylceramide in myelin. Biochem J. 2005;388:245–54. [PMC free article: PMC1186713] [PubMed: 15658937]
- Edvardson S, Hama H, Shaag A, Gomori JM, Berger I, Soffer D, Korman SH, Taustein I, Saada A, Elpeleg O. Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia. Am J Hum Genet. 2008;83:643–8. [PMC free article: PMC2668027] [PubMed: 19068277]
- Guo L, Zhou D, Pryse KM, Okunade AL, Su X. Fatty acid 2-hydroxylase mediates diffusional mobility of Raft-associated lipids, GLUT4 level, and lipogenesis in 3T3-L1 adipocytes. J Biol Chem. 2010;285:25438–47. [PMC free article: PMC2919107] [PubMed: 20519515]
- Justesen CR, Penn RD, Kroin JS, Egel RT. Stereotactic pallidotomy in a child with Hallervorden-Spatz disease. Case report. J Neurosurg. 1999;90:551–4. [PubMed: 10067928]
- Kruer MC, Paisán-Ruiz C, Boddaert N, Yoon MY, Hama H, Gregory A, Malandrini A, Woltjer RL, Munnich A, Gobin S, Polster BJ, Palmeri S, Edvardson S, Hardy J, Houlden H, Hayflick SJ. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann Neurol. 2010;68:611–8. [PMC free article: PMC6059612] [PubMed: 20853438]
- Levy M, Llinas RH. Deferiprone reduces hemosiderin deposits in the brain of a patient with superficial siderosis. AJNR Am J Neuroradiol. 2011;32:E1–2. [PMC free article: PMC7964941] [PubMed: 21051507]
- Magariello A, Russo C, Citrigno L, Zuchner S, Patitucci A, Mazzei R, Conforti FL, Ferlazzo E, Aguglia U, Muglia M. Exome sequencing reveals two FA2H mutations in a family with a complicated form of Hereditary Spastic Paraplegia and psychiatric impairments. J Neurol Sci. 2017;372:347–9. [PubMed: 28017243]
- Pierson TM, Simeonov DR, Sincan M, Adams DA, Markello T, Golas G, Fuentes-Fajardo K, Hansen NF, Cherukuri PF, Cruz P, Mullikin JC, Blackstone C, Tifft C, Boerkoel CF, Gahl WA., NISC Comparative Sequencing Program. Exome sequencing and SNP analysis detect novel compound heterozygosity in fatty acid hydroxylase-associated neurodegeneration. Eur J Hum Genet. 2012;20:476–9. [PMC free article: PMC3306865] [PubMed: 22146942]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Ristori G, Romano S, Visconti A, Cannoni S, Spadaro M, Frontali M, Pontieri FE, Vanacore N, Salvetti M. Riluzole in cerebellar ataxia: a randomized, double-blind, placebo-controlled pilot trial. Neurology. 2010;74:839–45. [PubMed: 20211908]
- Soehn AS, Rattay TW, Beck-Wödl S, Schäferhoff K, Monk D, Döbler-Neumann M, Hörtnagel K, Schlüter A, Ruiz M, Pujol A, Züchner S, Riess O, Schüle R, Bauer P, Schöls L. Uniparental disomy of chromosome 16 unmasks recessive mutations of FA2H/SPG35 in 4 families. Neurology. 2016;87:186–91. [PMC free article: PMC4940069] [PubMed: 27316240]
- Tonelli A, D’Angelo MG, Arrigoni F, Brighina E, Arnoldi A, Citterio A, Bresolin N, Bassi MT. Atypical adult onset complicated spastic paraparesis with thin corpus callosum in two patients carrying a novel FA2H mutation. Eur J Neurol. 2012;19:e127–9. [PubMed: 22925154]
- Vanderver A, Prust M, Tonduti D, Mochel F, Hussey HM, Helman G, Garbern J, Eichler F, Labauge P, Aubourg P, Rodriguez D, Patterson MC, Van Hove JL, Schmidt J, Wolf NI, Boespflug-Tanguy O, Schiffmann R, van der Knaap MS, et al. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol Genet Metab. 2015;114:494–500. [PMC free article: PMC4390457] [PubMed: 25649058]
- Zaki MS, Selim L, Mansour L, Mahmoud IG, Fenstermaker AG, Gabriel SB, Gleeson JG. Mutations in FA2H in three Arab families with a clinical spectrum of neurodegeneration and hereditary spastic paraparesis. Clin Genet. 2015;88:95–7. [PubMed: 25496456]
Publication Details
Author Information and Affiliations
Oregon Health & Science University
Portland, Oregon
Children's Hospital of Eastern Ontario
Ottawa, Ontario, Canada
Oregon Health & Science University
Portland, Oregon
Publication History
Initial Posting: June 28, 2011; Last Update: September 27, 2018.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Gregory A, Venkateswaran S, Hayflick SJ. Fatty Acid Hydroxylase-Associated Neurodegeneration. 2011 Jun 28 [Updated 2018 Sep 27]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
