Clinical Description
DYNC1H1-related disorders are primarily characterized by an axonal neuropathy with a wide phenotypic spectrum, ranging from a neuromuscular-only phenotype (DYNC1H1-related neuromuscular disorder) to phenotypes involving both the central nervous system and peripheral nervous system referred to collectively as DYNC1H1-related neurodevelopmental disorder [Amabile et al 2020, Becker et al 2020, Dafsari et al 2021].
DYNC1H1-related neuromuscular disorder (DYNC1H1-NMD). Manifestations are limited to the peripheral nervous system (PNS) and characterized predominantly by motor neuropathy, initially most pronounced in the lower limbs; muscle weakness and atrophy variably associated with foot deformities, contractures, and other skeletal involvement; and/or delayed motor milestones.
DYNC1H1-related neurodevelopmental disorder (DYNC1H1-NDD). Manifestations include motor axonal neuropathy and global developmental delay / intellectual disability, epilepsy, neurobehavioral/psychiatric manifestations, and movement disorders with or without malformations of cortical development (MCD) and or microcephaly. In an individual with more significant central nervous system (CNS) involvement, the motor axonal neuropathy may not be evident clinically and thus only detected on further evaluation such as electrophysiologic testing.
To date, more than 200 individuals with DYNC1H1-related disorders have been identified [Amabile et al 2020, Becker et al 2020]. The following comparison of the two phenotypic designations for DYNC1H1-related disorders is based on these reports (see and Table 2).
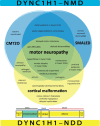
Venn diagram provides an overview of key phenotypes and clinical entities in DYNC1H1-related neuromuscular disorder (DYNC1H1-NMD; indicated in blue) and DYNC1H1-related neurodevelopmental disorder (DYNC1H1-NDD; indicated in yellow). Note that motor neuropathy (more...)
DYNC1H1-Related Neuromuscular Disorder (DYNC1H1-NMD)
Motor axonal neuropathy. In general, the proximal muscles of the lower limbs initially are most severely affected, with progression during childhood to the entire proximal lower limbs and subsequently during adolescence or adulthood to the upper limbs [Harms et al 2010, Tsurusaki et al 2012, Peeters et al 2015, Scoto et al 2015, Ding et al 2016, Hertecant et al 2016]. Up to 95% of individuals have reduced lower limb muscle strength and about 20% have reduced upper limb strength [Becker et al 2020].
Initial findings of lower limb involvement include decreased muscle tone and weakness due to muscle atrophy and reduced muscle mass. In some infants, decreased fetal movements result in secondary contractures evident at birth [Scoto et al 2015, Becker et al 2020], including the following:
Foot deformities, present in around 50% of individuals, such as pes cavus (in around half of all cases), pes equinus, pes equinovarus, pes adductus, talus verticalis, shortened forefoot, slender/hammer toes, pes calcaneus, hyperextension deformities, and bilateral foot drop [
Weedon et al 2011,
Beecroft et al 2017,
Chen et al 2017,
Chan et al 2018,
Becker et al 2020,
Liu et al 2023]
Other contractures, present in around 10% of individuals, almost exclusively affecting the lower limb, including the hips, iliotibial ligament, knees, and Achilles tendon; can also involve the upper limb (especially the thumbs) [
Scoto et al 2015,
Amabile et al 2020,
Becker et al 2020]. Congenital unilateral or bilateral hip dysplasia and/or dislocation, a relatively rare manifestation, may be evident prenatally on ultrasound examination or after birth [
Scoto et al 2015].
Sensory involvement, especially of the lower limbs, can include transient paresthesias, neuropathic pain, reduced or lost proprioception, and reduced response to pinprick, fine touch, and/or vibration. Age-related progression may manifest during adulthood as leg fatigue and pain [Harms et al 2010].
Motor development is moderately to severely delayed in about 50% of individuals with DYNC1H1-NMD [Becker et al 2020, Dafsari et al 2021]. An abnormal waddling gait, present in 30% of individuals due to reduced proprioception and muscle weakness in the proximal lower limbs, is characterized by minor imbalance or recurrent falls and difficulties in running [Weedon et al 2011, Niu et al 2015, Scoto et al 2015, Becker et al 2020].
As neuropathy progresses, the gait may become ataxic and walking aids such as canes or wheelchairs may be required [Chan et al 2018].
Other less common motor involvement includes the following:
Histological findings. Click here (pdf) for histologic findings in PNS studies.
DYNC1H1-Related Neurodevelopmental Disorder (DYNC1H1-NDD)
Individuals with DYNC1H1-NDD exhibit the motor axonal neuropathy described previously for DYNC1H1-NMD, as well as additional variable features.
Neurodevelopmental delays including motor, speech, and/or cognitive development may occur [Weedon et al 2011, Becker et al 2020]. Developmental regression in these three domains has been reported in a few individuals [Amabile et al 2020, Yang et al 2021].
Neurobehavioral/psychiatric manifestations affect more than 15% of individuals with DYNC1H1-NDD [Becker et al 2020]. The most common is attention-deficit hyperactivity disorder. Other behavioral disorders include attention disorders without hyperactivity, dyslexia, autism spectrum disorder, aggressive behavior, and motor stereotypies [Chan et al 2018, Amabile et al 2020, Becker et al 2020].
Epilepsy. Seizures, reported in nearly 40% of individuals, are infantile onset in about 60% of all individuals with seizures [Becker et al 2020, Liu et al 2023]. The most common seizure types are focal (27%), followed by generalized onset (10%) and mixed focal and generalized onset (8%) [Chung et al 2022]. Infantile epileptic spasms syndrome (IESS) was reported in over 10% of individuals with seizures [Yang et al 2021, Su et al 2022, Liu et al 2023].
Other reported specific electroclinical syndromes are centrotemporal epilepsy, acquired aphasia syndrome, focal epilepsy of structural origin, and Lennox-Gastaut syndrome [Liu et al 2023].
Electroencephalography (EEG) may show different patterns according to seizure semiology, including the following:
Movement disorders. Parkinsonism, reported in one individual with global developmental delay, extrapyramidal findings (bradykinesia, hypokinesia, cogwheel rigidity, small step walking, difficulty in initiating movements, hypomimic face, and a resting tremor), responded favorably to treatment with levodopa [Szczałuba et al 2018].
Limb ataxia has been rarely reported [Strickland et al 2015, Fernández Perrone et al 2022].
Vocal cord paresis and dysarthria have been rarely reported and may be different from the speech disorders associated with neurodevelopmental delay [Jamuar et al 2014, Zillhardt et al 2016, Amabile et al 2020].
Brain MRI abnormalities have been detected on in more than 60% of individuals studied and include the following [Poirier et al 2013, Fiorillo et al 2014, Jamuar et al 2014, Scoto et al 2015, Gelineau-Morel et al 2016, Hertecant et al 2016, Su et al 2022, Liu et al 2023]:
Malformations of cortical development including pachygyria (that may resemble lissencephaly), polymicrogyria, a much broader range of cortical dysgyria (48%), and dysgenesis or agenesis of the corpus callosum (23%)
Ventriculomegaly (15%)
Cerebellar hypoplasia (14%)
Gray matter heterotopia (11%)
Brain stem hypoplasia (9%)
Other findings seen in one or a few individuals include abnormalities of the white matter [Gelineau-Morel et al 2016, Chan et al 2018, Liu et al 2023], cortical atrophy [Becker et al 2020], dysmorphic basal ganglia [Poirier et al 2013], schizencephaly [Liu et al 2023], arachnoid cyst [Chan et al 2018], large cerebrospinal fluid spaces [Hertecant et al 2016], and hydrocephalus [Scoto et al 2015].
Microcephaly, affecting about 5% of individuals, is most often congenital [Poirier et al 2013, Laquerriere et al 2017]. In other individuals, head circumference may be normal at birth, with microcephaly becoming evident postnatally [Hertecant et al 2016, Laquerriere et al 2017, Becker et al 2020].
Neuropathologic examination. Click here (pdf) for findings from neuropathologic examinations.
Other Findings
The following findings have been observed across the entire phenotypic spectrum of DYNC1H1-related disorders:
Congenital anomalies of kidney and urinary tract (CAKUT), reported in <5% of individuals, including dilatated uropathy [
Becker et al 2020]
Other manifestations, including intermittent painful muscle cramps with moderate exercise [
Beecroft et al 2017], intrauterine growth restriction, hydrops fetalis [
Zillhardt et al 2016], osteocutaneous anomalies (e.g., prominent calcanei, cutis laxa), accessory spleen, congenital anterior diaphragmatic hernia, syringomyelia [
Becker et al 2020], cryptorchidism [
Minardi et al 2020], hypospadias [
Amabile et al 2020], celiac disease, and eosinophilic esophagitis [
Fernández Perrone et al 2022].
Cardiovascular manifestations, perhaps incidental due to their prevalence in the general population, were reported in <5% of
DYNC1H1-related disorders and include mild aortic valve insufficiency, bicuspid aortic valve, anomalous pulmonary venous drainage, and atrial septum defect resulting in ventricular hypertrophy [
Amabile et al 2020,
Becker et al 2020,
Liu et al 2023].
Genotype-Phenotype Correlations
Several genotype-phenotype correlations have been observed based on the location of pathogenic variants within the four main functional domains of DYNC1H1 [Becker et al 2020, Dafsari et al 2021] (see ); however, limited data to date prevent making more precise determinations.
Beginning tail (comprising amino acids 1-299 and 1141-1373). Pathogenic variants in this region are mainly associated with DYNC1H1-NDD and behavioral abnormalities.
Dimerization domain (comprising amino acids 300-1140). Pathogenic variants in this region lead to generally milder, primarily neuromuscular disorders (DYNC1H1-NMD).
Linker domain (comprising amino acids 1374-1867). Pathogenic variants in this region are particularly associated with DYNC1H1-NDD and behavioral abnormalities.
Motor domain (comprising amino acids 1868-4221). Pathogenic variants in this domain have been found mainly in individuals with DYNC1H1-NDD with malformations of cortical development, epilepsy, and neurobehavioral/psychiatric manifestations [Amabile et al 2020, Becker et al 2020, Chung et al 2022].