Summary
Clinical characteristics.
Spondylocostal dysostosis (SCDO), defined radiographically as multiple segmentation defects of the vertebrae in combination with abnormalities of the ribs, is characterized clinically by a short trunk in proportion to height; short neck; and non-progressive mild scoliosis in most affected individuals – rarely, more significant scoliosis occurs. Respiratory function in neonates with severe disease may be compromised by reduced size of the thorax. By age two years lung growth may improve sufficiently to support relatively normal growth and development. In severely affected individuals with restricted pulmonary capacity, there is a possibility that pulmonary hypertension may eventually impact cardiac function. Males with SCDO appear to be at increased risk for inguinal hernia.
Diagnosis/testing.
The diagnosis of SCDO is based on radiographic features. Identification of biallelic pathogenic variants in DLL3, HES7, LFNG, MESP2, RIPPLY2, or TBX6 can confirm the diagnosis of autosomal recessive SCDO.
Management.
Treatment of manifestations: Surgical intervention may be necessary when scoliosis is significant; external bracing (e.g., by use of a vertical expandable prosthetic titanium rib) may be considered, as well as growing rods and other devices as appropriate. Respiratory support, including intensive care, is provided as needed for the small proportion of individuals with acute respiratory distress and chronic respiratory failure. Expert management is indicated for chronic respiratory failure, which can result in pulmonary hypertension and cardiac failure. Standard treatment of neurologic problems associated with LFNG-related SCDO. Inguinal hernias are treated per routine.
Surveillance: Growth, spinal curvature, respiratory function, neurologic and motor function, and development should be monitored. The parents / care providers of young males need to be alert for the signs of inguinal hernia and its potential complications.
Genetic counseling.
SCDO caused by biallelic pathogenic variants in DLL3, HES7, LFNG, MESP2, RIPPLY2, or TBX6 is inherited in an autosomal recessive manner. (Autosomal dominant inheritance of TBX6-related SCDO has been reported in a three-generation family.) If both parents are known to be heterozygous for an autosomal recessive SCDO-causing pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the autosomal recessive SCDO-related pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible. In experienced hands, detailed fetal ultrasound scanning is sensitive enough to detect multiple segmentation defects of the vertebrae as early as 13 weeks' gestation, especially when the malformation is anticipated and looked for specifically. However, molecular genetic testing of an at-risk pregnancy is considered the gold standard for accurate prenatal diagnosis.
Diagnosis
Suggestive Findings
Spondylocostal dysostosis (SCDO) should be suspected in individuals with the following radiographic features and family history:
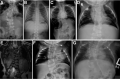
- Multiple segmentation defects of the vertebrae (M-SDV) most evident on anteroposterior radiograph of the whole spine. Abnormal segmentation of at least ten contiguous vertebrae. In the affected fetus or young child each vertebra is round or ovoid with smooth boundaries; the appearance of the vertebral column has been referred to as the "pebble beach" sign [Turnpenny et al 2003] (see Figure 1), especially in DLL3-related SCDO. As ossification proceeds after mid- to late childhood, the "pebble beach" appearance gives way to multiple irregularly shaped vertebral bodies and hemivertebrae that may be difficult to distinguish individually on plain x-ray.
- Mild scoliosis
- Rib abnormalities. Malalignment of at least some ribs with a variable number of intercostal rib fusions, and sometimes a reduction in rib number
- No major asymmetry to the shape of the thorax
- Family history consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.

Establishing the Diagnosis
The diagnosis of autosomal recessive SCDO is established in a proband with suggestive radiographic features and biallelic pathogenic (or likely pathogenic) variants identified by molecular genetic testing in one of the genes listed in Table 1.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include likely pathogenic variants. (2) Identification of biallelic variants of uncertain significance (or of one known pathogenic variant and one variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel, serial single-gene testing), and comprehensive genomic testing (exome sequencing, genome sequencing). Gene-targeted testing requires that the clinician determine which gene(s) are likely involved (see Option 1), whereas comprehensive genomic testing does not (see Option 2).
Option 1
A multigene panel that includes DLL3, HES7, LFNG, MESP2, RIPPLY2, TBX6, and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Serial single-gene testing. Prioritized genetic testing may be pursued as single-gene testing based on clinical features:
- Sequence analysis of LFNG can be performed first in individuals with severe truncal shortening observed on radiographs.
- Sequence analysis of MESP2 can be performed first in individuals with radiographic features more typical of spondylothoracic dysplasia (or dysostosis) (STD); the ribs tend to be straight and more regularly aligned than in the other forms of SCDO (i.e., demonstrating fewer points of fusion), resulting in a "crab-like" appearance.
Option 2
When the phenotype is indistinguishable from many other skeletal dysplasias, comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Autosomal Recessive Spondylocostal Dysostosis
Clinical Characteristics
Clinical Description
Spondylocostal dysostosis (SCDO), defined radiographically as multiple segmentation defects of the vertebrae that is usually generalized throughout the spine, is characterized clinically by a short trunk in proportion to height, short neck, and non-progressive mild scoliosis in most affected individuals. To date, nearly 100 individuals have been identified and/or reported with SCDO and biallelic pathogenic variants in one of the genes listed in Table 1. The following description of the phenotypic features associated with this condition is based on the cited reports.
Skeletal. Multiple segmentation defects of the vertebrae, which is usually generalized throughout the spine, results in:
- A short trunk in proportion to height. The extent varies and the data is very limited, but based on leg length measurements, individuals with SCDO are 10% shorter than projected adult height. Some individuals have severe short stature, with height up to four standard deviations below the mean [Sparrow et al 2006; Schuhmann et al 2021; P Turnpenny, personal communication]. LNFG-related SCDO appears to be associated with shorter stature compared to other causes of SCDO (see Figure 2). The reason(s) for this variability is not well understood.
- Short neck. The extent varies, and the data is limited, but – similar to the decrease in overall spine length – the neck is likely to be shortened by approximately 10%. The range of limitation in neck mobility has not been formally assessed.
- Non-progressive, or mildly progressive but self-limiting, scoliosis occurs in most affected individuals, usually apparent radiographically in infancy. More significant scoliosis, with a greater degree of progression, especially at the thoracolumbar region, is apparent in individuals with LFNG-related SCDO [Sparrow et al 2006, Takeda et al 2018, Schuhmann et al 2021]. Severe scoliosis, including the need for scoliosis surgery, appears to be relatively rare [A Cornier, personal communication; P Turnpenny, personal communication].
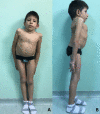
Figure 2.
Individual with LFNG-related SCDO at age seven years nine months showing marked truncal shortening Courtesy of Prof Eleni Fryssira, Athens, Greece. LFNG analysis performed in Lausanne, Switzerland.
Respiratory. The most important consideration in neonates diagnosed with SCDO is impaired respiratory function, which may be compromised by reduced size of the thorax. In these infants, respiratory insufficiency may be the presenting clinical problem. Life-threatening respiratory insufficiency requiring neonatal intensive care appears to be rare but anecdotally has been known to occur. One individual died in infancy from respiratory insufficiency and at postmortem was found to have a membranous left hemidiaphragm [Turnpenny et al 1999]. This has not been reported in any other individuals with SCDO. The most significant potential secondary complication is chronic respiratory failure caused by reduced lung capacity in individuals with severe disease.
In children requiring early respiratory support, lung growth may improve sufficiently to support relatively normal growth and development by age two years. However, life-threatening complications can occur, especially pulmonary hypertension and cardiac failure in individuals with severely restricted lung capacity from birth. There is no systematic review concerning susceptibility to pulmonary infection and pneumonia or incidence of pulmonary hypertension.
Inguinal hernia. Males with SCDO appear to be at increased risk for inguinal hernia, which has been noted in the neonatal period [Turnpenny et al 1999, Turnpenny et al 2003, Otomo et al 2019].
Neurologic complications appear to be rare. Lumbosacral meningomyelocele was reported in one individual [Sparrow et al 2008] and a neural tube defect occurred in a second individual [Authors, personal communication] with HES7-related SCDO. Syringomyelia was identified at age seven years in one individual with LFNG-related SCDO on spine MRI performed due to new balance problems. By age ten years this individual had urinary incontinence; he was intellectually normal [E Fryssira and M Christodoulou, personal communication]. Distal arthrogryposis was reported in one individual with LFNG-related SCDO; it was not clear whether this was a primary abnormality or secondary to impingement of neural pathways in the cervical vertebrae [Sparrow et al 2006].
Other
- Cosegregation of dextrocardia was reported in a large consanguineous Middle Eastern kindred with HES7-related SCDO (see Figure 3) [Sparrow et al 2013a]; whether this was due to HES7-related SCDO or a separate genetic cause has not been established.
- Solitary pelvic kidney, uterine dysgenesis, absence epilepsy, and inner ear (presumed sensorineural) deafness were reported in one individual with LFNG-related SCDO [Schuhmann et al 2021].
Prognosis. In the absence of restricted lung capacity, individuals with SCDO have normal life expectancy. The risk of pulmonary hypertension and associated complications is unknown.
Phenotype Correlations by Gene
DLL3. Scoliosis is generally mild and non-progressive, and the need for surgical intervention to stabilize the spine is rare. However, more significant scoliosis has been observed in some individuals (see Figure 4) [A Cornier, personal communication].

Figure 4.
Radiograph of infant with DLL3-related SCDO and unusually severe scoliosis
HES7. Radiographic features in the limited number of published individuals have ranged from resembling spondylothoracic dysostosis (STD) [Sparrow et al 2008] (see Figure 5) to those typical in DLL3-related SCDO; all vertebrae display abnormal segmentation.
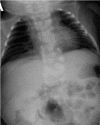
LFNG. Shortening of the spine and scoliosis appear to be more severe in individuals with LFNG-related SCDO compared to that seen in DLL3-, HES7-, and MESP2-related SCDO, because all vertebral bodies appear to show more severe segmentation defects (see Figure 2, Figure 6, and Figure 7) [Lefebvre et al 2018]. Rib anomalies are similar to those seen in DLL3- and MESP2-related SCDO.
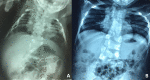
Figure 6.
Radiographs of a child with LFNG-related SCDO A. Spine radiograph as a neonate. The pattern of malsegmentation is not clearly distinguishable from typical findings in DLL3-related SCDO.
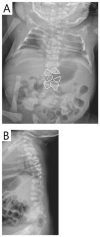
Figure 7.
Segmentation defects of the vertebrae of the entire spine with angulated vertebral bodies (dotted lines) at birth in an individual with LNFG-related SCDO (patient 18 of Lefebvre et al [2018]) Reprinted with permission from Lefebvre et al [2018]
MESP2. Spine radiographs in individuals with MESP2-related SCDO show at least some disruption of all vertebral segments. However, lumbar vertebrae are relatively mildly affected compared to thoracic vertebrae (see Figure 8). In the limited reports thus far, the ribs tend to be straight and more regularly aligned than in other forms of SCDO (i.e., demonstrating fewer points of fusion).
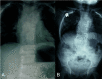
Figure 8.
Radiographs of a child with MESP2-related SCDO. The generalized segmentation defects of the vertebrae show more angular features than is typical of DLL3-related SCDO.
RIPPLY2. Two brothers with RIPPLY2-related SCDO had vertebral segmentation defects affecting the posterior elements of C1-C4 and hemivertebrae and butterfly vertebrae of T2-T7 (see Figure 9). Marked cervical kyphosis at C2-C3 was associated with cord compression, and mild thoracic scoliosis was present [McInerney-Leo et al 2015]. Three individuals from two families, with the same pathogenic variant [Serey-Gaut et al 2020], had agenesis of the posterior and lateral elements of most cervical vertebrae, with limited and variable involvement of some thoracic vertebrae. The radiologic pattern was distinct from other forms of SCDO, and RIPPLY2-related SCDO may be better categorized as a form of Klippel-Feil anomaly.
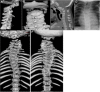
TBX6. Although the number of reported individuals with TBX6-related SCDO is limited, this phenotype resembles that of DLL3-related SCDO. Radiologically it is almost indistinguishable (see Figure 10) [C Shaw-Smith, personal communication].
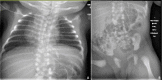
Figure 10.
Thoracic (A) and lumbar spine (B) radiographs of an infant with TBX6-related SCDO Courtesy of Charles Shaw-Smith, Exeter, UK
See Figure 11 for radiographic comparison of DLL3-, LFNG-, HES7-, and TBX6-related SCDO and MESP2-related STD.

Figure 11.
Radiologic features for the different genes identified in a cohort of individuals with regional multiple segmentation defects of the vertebrae A-D. An individual with DLL3-related SCDO
Genotype-Phenotype Correlations
DLL3. The radiographic features of DLL3-related SCDO appear to be very consistent (see Figure 1). However, two individuals homozygous for DLL3 pathogenic missense variants in the region encoding the EGF domain had slightly milder phenotypes (see Figure 12). Some evidence suggests that these pathogenic missense variants would allow the EGF domains to adopt the correct fold in the DLL3 protein but perhaps be thermodynamically less stable than the wild type protein [Authors, unpublished data]. However, some of the pathogenic missense variants identified in affected individuals cause a phenotype that is indistinguishable from that caused by DLL3 pathogenic truncating variants. This probably results from the different effects conferred upon protein folding compared to those pathogenic missense variants associated with the slightly milder phenotype.
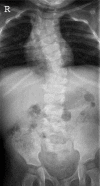
Figure 12.
Radiograph of a child with a mild form of DLL3-related SCDO. All vertebrae show at least some relatively mild segmentation abnormality.
MESP2. The 4-bp duplication c.500_503dup occurs after the basic helix-loop-helix (bHLH) domain and causes a frameshift resulting in a premature stop codon within the second (and final) MESP2 exon [Whittock et al 2004b]. Transcripts with this pathogenic variant would not be subject to nonsense-mediated decay. Individuals with this pathogenic variant are predicted to have a truncated protein containing an intact bHLH domain, which may retain some function. In contrast, the pathogenic nonsense variants identified in spondylothoracic dysostosis (STD) (see Genetically Related Disorders) are located within the first exon, and the resulting mutated mRNA transcripts are predicted to be susceptible to nonsense-mediated decay. Therefore, persons homozygous or compound heterozygous for these pathogenic nonsense variants are likely to have reduced or absent levels of MESP2 protein, which may account for the difference in severity between the MESP2-related SCDO and STD phenotypes.
TBX6. See Genetically Related Disorders for genotype-phenotype correlations observed in allelic disorders.
No genotype-phenotype correlations for HES7, LFNG, or RIPPLY2 have been identified.
Penetrance
To date, penetrance appears to be complete for the pathogenic variants implicated in autosomal recessive SCDO.
Nomenclature
The term Jarcho-Levin syndrome (JLS) [Jarcho & Levin 1938] has been used (confusingly) to refer to:
- All radiologic phenotypes that include segmentation defects of the vertebrae (SDV) and abnormal rib alignment, including reports of phenotypes that are neither similar to the case description of Jarcho & Levin [1938] nor consistent with spondylothoracic dysostosis (STD);
- STD in Puerto Ricans of Spanish descent (see Genetically Related Disorders).
Use of the terms costovertebral dysplasia and spondylothoracic dysostosis/dysplasia for segmentation abnormalities of the spine and ribs has led to great confusion. Note: These disorders are dysostoses rather than dysplasias:
- "Costovertebral dysplasia" is now used less frequently.
- "Spondylothoracic dysostosis/dysplasia" (STD) is recognized as being distinct from SCDO (see Differential Diagnosis).
- Note: Spondylothoracic dysostosis is referred to as "vertebral segmentation defect (congenital scoliosis) with variable penetrance" in the 2023 revision of the Nosology of Genetic Skeletal Disorders [Unger et al 2023]. It is the authors' judgment that STD does not fit within the broad category of "congenital scoliosis." Congenital scoliosis is usually caused by segmentation defects in a single vertebra (or segmentation defects in a limited number of vertebrae within one region of the spine), whereas STD is a generalized form of SDV, and scoliosis is not a major feature.
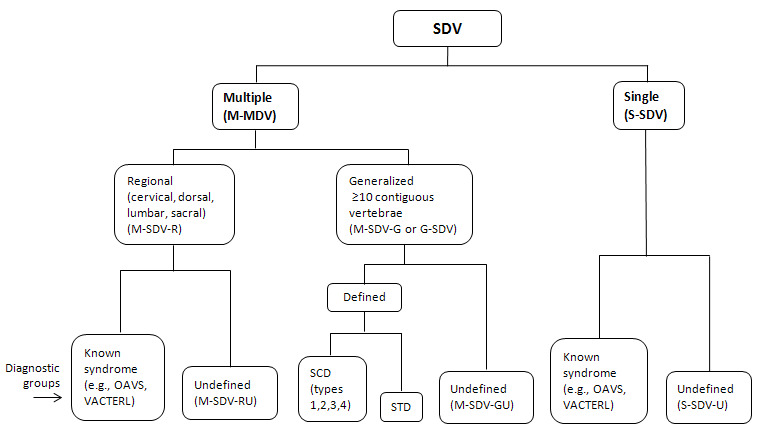
The wide range of radiologic phenotypes with multiple segmentation defects of the vertebrae (M-SDV) within SCDO has highlighted the need to rationalize nomenclature for these diverse and poorly understood disorders. The International Consortium for Vertebral Anomalies and Scoliosis (ICVAS), now subsumed into the International Consortium for Scoliosis Genetics Development and Disease (ICSGDD), proposed two algorithms:
- The clinical algorithm, used for routine reporting of SDV, identifies seven broad categories (see Figure 13). For the purposes of clinical reporting, additional comments can describe SDV findings in more detail.
- The research algorithm, used for more detailed documentation of SDV, employs ontology applicable to humans and animal models (see Figure 14).
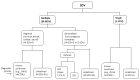
Figure 13.
ICVAS clinical classification algorithm All forms of SDV can be placed in one of seven broad categories. The classification combines a descriptive approach for the diverse radiologic phenotypes encountered in clinical practice with specific diagnoses (more...)
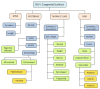
Figure 14.
ICVAS research classification algorithm: a more detailed, systematic analysis of radiographic anatomic features. Documentation of phenotypes in a systematic ontology facilitates direct interspecies comparison and stratification of patient cohorts for (more...)
Note: In the classification system proposed by the ICVAS, SCDO is the preferred term for generalized segmentation defects of the vertebrae (G-SDV) with rib involvement [Turnpenny et al 2007, Offiah et al 2010].
Klippel-Feil anomaly (KFA) refers to cervical vertebral fusion anomalies. The term "KFA" is used broadly for a number of phenotypes.
Prevalence
DLL3-related SCDO. Seventy-five percent of individuals have been the offspring of consanguineous unions (Exeter Laboratory experience), mostly of Middle Eastern or Pakistani origin, and occasionally of European origin and elsewhere. A small number of individuals from northern Europe (England, Wales, the Netherlands, and Switzerland) have been shown to be compound heterozygotes [Bonafé et al 2003, Whittock et al 2004a]. Assuming a period of time during which approximately one million births occurred, the carrier frequency in the European population in the UK would be approximately 1:350.
HES7-, LFNG-, MESP2-, and RIPPLY2-related SCDO have been reported in only a small number of individuals [Whittock et al 2004b, Bonafé & Superti-Furga 2005, Sparrow et al 2006, Sparrow et al 2008, Sparrow et al 2010, Sparrow et al 2013a, Lefebvre et al 2017]. TBX6-related SCDO has been reported more often, suggesting it is the second most common form of SCDO after DLL3-related SCDO.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in DLL3, HES7, LFNG, or RIPPLY2.
MESP2-related spondylothoracic dysostosis (MESP2-STD). To date, most individuals reported with MESP2-related STD have had pathogenic nonsense variants in exon 1 of MESP2, which are predicted to result in nonsense-mediated decay; however, several affected individuals have been reported who are heterozygous for a pathogenic nonsense variant and a pathogenic missense variant [Cornier et al 2008]. STD occurs most frequently in Puerto Ricans of Spanish descent [A Cornier, personal communication], presumably as a result of the MESP2 founder variant p.Glu103Ter (see Figure 15).
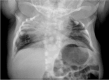
Figure 15.
Radiograph of an infant with MESP2-related STD. The spine is severely shortened with fusion of the ribs posteriorly at the costovertebral junctions. The ribs fan out in a "crab-like" manner, and in many individuals the ribs show no points of intercostal (more...)
TBX6. To date, TBX6 pathogenic variants have been associated with the following:
- Heterozygous TBX6 deletions, sometimes in trans with a hypomorphic allele, have been reported in individuals with congenital scoliosis and müllerian aplasia or renal malformations [Sandbacka et al 2013, Wu et al 2015, Li et al 2022]. The most commonly reported hypomorphic allele is the T-C-A permissive haplotype, defined by single-nucleotide polymorphisms at the positions of rs2289292, rs3809624, and rs38090627 [Chen et al 2020].
- Autosomal dominant spondylocostal dysostosis (SCDO) was identified in a three-generation family. The widespread vertebral malformations consisted of a mixture of hemivertebrae and blocks of fused segments. There was relative sparing of rib involvement. Mild scoliosis was centered on the mid-thoracic region. No other anomalies were identified, and neurodevelopment was normal. A heterozygous TBX6 pathogenic variant (p.Ter437CextTer81), disrupting the natural stop codon, segregated with the condition, and functional studies demonstrated approximately half the wild type transcriptional activation activity [Gucev et al 2010, Sparrow et al 2013b] (see Figure 8). Subsequent analysis showed that this TBX6 stop-lost variant was located within the T-C-A permissive haplotype. The combined reduction in TBX6 expression and transcriptional activation might therefore account for the SCDO phenotype or, possibly, the 81 amino acids added to the C-terminal may affect the ability of TBX6 to interact with other proteins that are critical in vertebral formation [Wu et al 2015].
- Variable segmentation anomalies, most commonly affecting the lower thoracic or upper lumbar regions and usually presenting as congenital scoliosis, have been reported. This may be due to deletion of TBX6 on one allele and a high-risk haplotype of polymorphisms on the other allele [Wu et al 2015, Takeda et al 2017].
- A distinct, very severe, lethal form of STD with müllerian duct anomalies was reported [P Turnpenny, unpublished data], more severe than the individual with severe TBX6-related SCDO reported by Errichiello et al [2020].
- Müllerian aplasia / MURCS association / Mayer-Rokitansky-Küster-Hauser syndrome has been reported [Sandbacka et al 2013].
Contiguous gene deletions. It has been determined that vertebral abnormalities and scoliosis observed in some individuals with the 16p11.2 recurrent deletion result from a combination of a TBX6 null allele (i.e., the recurrent deletion that encompasses TBX6 as well as multiple adjacent genes) and a hypomorphic TBX6 allele [Wu et al 2015]. The 16p11.2 recurrent deletion phenotype is characterized by motor speech disorder, language disorder, motor coordination difficulties, psychiatric conditions, and autistic features.
Differential Diagnosis
Rarely, spondylocostal dysostosis (SCDO) occurs in association with chromosome abnormalities; however, apart from trisomy 8 mosaicism, no consistent genomic region has been involved, and the significance of these associations is unknown.
Autosomal dominant SCDO. One family with autosomal dominant SCDO due to a heterozygous TBX6 pathogenic variant has been reported (OMIM 122600). Additional families with autosomal dominant SCDO without an identified gene have also been reported; in these families the extent of segmentation defects of the vertebrae is quite variable [Rimoin et al 1968, Kubryk & Borde 1981, Temple et al 1988, Lorenz & Rupprecht 1990].
Spondylothoracic dysostosis (STD), despite similarities to autosomal recessive SCDO, has distinctive phenotypic features that warrant this separate designation. Infants with STD are at the highest risk for respiratory insufficiency and have a nearly 50% mortality rate by the end of infancy [Cornier et al 2004]. To date, most individuals reported with STD have had pathogenic nonsense variants in exon 1 of MESP2 (see Genetically Related Disorders). The differences in the radiographic findings in STD that distinguish it from SCDO include the following (see Figure 11):
- More severe shortening of the spine (all vertebral segments affected), especially the thoracic spine, leading to impaired respiratory function in infancy
- Rib fusions typically occurring posteriorly at the costovertebral origins, where the spinal shortening is most severe. The ribs usually appear straight and neatly aligned without points of fusion along their length. On anteroposterior x-ray the ribs characteristically "fan out" from their costovertebral origins in a "crab-like" fashion.
- A distinctive radiographic appearance called the "tramline sign" that results from early radiographic prominence of the vertebral pedicles, in contrast to the vertebral bodies, which have no regular form or layout [Turnpenny et al 2007].
Segmentation defects of the vertebrae (SDV) are estimated to occur in 0.5-1.0 in 1,000 live births, but in clinical practice the radiologic phenotypes and syndromic associations are extremely diverse. Syndromic forms of multiple segmentation defects of the vertebrae (M-SDV) should be considered if the diagnostic criteria for SCDO or STD are not met. For most individuals the underlying cause is not known, but an increasing number of genes are being identified. Some of the M-SDV syndromes to consider are listed in Tables 2a and 2b.
Table 2a.
Selected Genes Associated With M-SDV (SCDO and STD excluded)
Table 2b.
Other Syndromes/Conditions That Include M-SDV (SCDO and STD excluded)
Note: A single individual with an SCDO-like phenotype with multiple regional segmentation defects of the vertebrae, multiple intervertebral fusions of laminae, dysmorphic features, and cleft palate has been reported in association with homozygosity for a start-loss variant in DMRT2 [Bouman et al 2018]. With severe left-sided rib cage deficiency, the infant died at age nine days. It is not yet known if DMRT2-related SCDO is a distinct entity.
Neural tube defects are also frequently associated with adjacent severe segmentation anomalies of one or more vertebrae. However, current consensus is that the diagnosis of SCDO should be reserved for individuals with abnormal segmentation of at least ten contiguous vertebrae.
Management
No clinical practice guidelines for autosomal recessive spondylocostal dysostosis (SCDO) have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with autosomal recessive SCDO, the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Autosomal Recessive Spondylocostal Dysostosis: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
In the majority of individuals, treatment is conservative because the clinical manifestations of the vertebral and rib malformations do not increase with age.
Table 4.
Autosomal Recessive Spondylocostal Dysostosis: Treatment of Manifestations
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 5 are recommended.
Table 5.
Autosomal Recessive Spondylocostal Dysostosis: Recommended Surveillance
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Virtually all individuals with SCDO have relative truncal shortening, and some have generalized short stature. For affected women, pregnancy may give rise to exaggerated intra-abdominal pressure problems, though there is no published research on this issue. As the spine is distorted, there are likely to be concerns with offering spinal and/or epidural anesthesia. However, spinal anesthesia has been successfully administered [Dolak & Tartt 2009].
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Spondylocostal dysostosis (SCDO) caused by biallelic pathogenic variants in DLL3, HES7, LFNG, MESP2, RIPPLY2, or TBX6 is inherited in an autosomal recessive manner.
Note: Autosomal dominant inheritance of TBX6-related SCDO has been reported in a three-generation family (all affected family members were male) [Sparrow et al 2013b]. Autosomal dominant inheritance is not discussed further in this section.
Pseudodominant inheritance. Although rare, there have been reports of SCDO appearing to be inherited in an autosomal dominant manner, although the extent of segmentation defects of the vertebrae (SDV) is variable [Temple et al 1988, Gucev et al 2010, Sparrow et al 2012]. In one such family [Floor et al 1989] the inheritance pattern was shown to be an example of pseudodominant inheritance (i.e., an autosomal recessive condition present in individuals in two or more generations of a family, thereby appearing to follow a dominant inheritance pattern) of DLL3-related SCDO in a highly consanguineous family [Turnpenny et al 1999, Whittock et al 2004a].
Risk to Family Members (Autosomal Recessive Inheritance)
Parents of a proband
- The parents of an affected child are presumed to be heterozygous for an autosomal recessive SCDO-causing pathogenic variant.
- If a molecular diagnosis has been established in the proband, molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for an autosomal recessive SCDO-causing pathogenic variant and to allow reliable recurrence risk assessment.
- If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, it is possible that one of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017]. If the proband appears to have homozygous pathogenic variants (i.e., the same two pathogenic variants), additional possibilities to consider include:
- A single- or multiexon deletion in the proband that was not detected by sequence analysis and that resulted in the artifactual appearance of homozygosity;
- Uniparental isodisomy for the parental chromosome with the pathogenic variant that resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for an autosomal recessive SCDO-causing pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. Unless an affected individual's reproductive partner* has heterozygous or biallelic pathogenic variants in the same autosomal recessive SCDO-related gene as that involved in the proband, offspring will be obligate heterozygotes (carriers) for an autosomal recessive SCDO-causing pathogenic variant.
* Molecular genetic testing for reproductive partners is appropriate, particularly if consanguinity is likely. Approximately 75% of individuals with autosomal recessive SCDO are from consanguineous families, usually from communities in which cousin partnerships are common.
Other family members. Each sib of the proband's parents is at 50% risk of being a carrier of an autosomal recessive SCDO-causing pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the autosomal recessive SCDO-causing pathogenic variants in the family. See Related Genetic Counseling Issues, Family planning.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
- Molecular genetic carrier testing of individuals from high-risk families, in which one or more individuals has been diagnosed with SCDO, may be helpful in identifying at-risk couples.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the autosomal recessive SCDO-related pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Fetal ultrasound examination. In experienced hands, detailed fetal ultrasound scanning is sensitive enough to detect multiple segmentation defects of the vertebrae (M-SDV) as early as 13 weeks' gestation, especially when the malformation is anticipated and looked for specifically. However, molecular genetic testing of an at-risk pregnancy is considered the gold standard for accurate prenatal diagnosis. Note: Gestational age is expressed as menstrual weeks calculated either from the first day of the last normal menstrual period or by ultrasound measurements.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- UCLA International Skeletal Dysplasia Registry (ISDR)Phone: 310-825-8998
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Spondylocostal Dysostosis, Autosomal Recessive: Genes and Databases
Table B.
OMIM Entries for Spondylocostal Dysostosis, Autosomal Recessive (View All in OMIM)
Molecular Pathogenesis
The six genes known to be associated with the six subtypes of autosomal recessive spondylocostal dysostosis (SCDO) encode proteins that are key components of the Notch signaling pathway, which (together with FGF and Wnt signaling) is one of the developmental pathways essential to normal somitogenesis [Dequéant et al 2006, Dequéant & Pourquié 2008].
- DLL3 encodes a ligand of NOTCH1 that inhibits signaling.
- HES7 encodes a basic helix-loop-helix (bHLH)-orange domain transcriptional repressor protein. HES7 is a direct target of NOTCH1 receptor signaling and is also a cycling gene expressed in the presomitic mesoderm.
- LFNG encodes a glycosyltransferase that post-translationally modifies the Notch family of cell-surface receptors. LFNG is a direct target of NOTCH1 receptor signaling and is also a cycling gene expressed in the presomitic mesoderm.
- MESP2 encodes a member of the bHLH family of transcriptional regulatory proteins. MESP2 is a direct target of NOTCH1 receptor signaling.
- RIPPLY2 is a negative regulator of TBX6 and is a direct transcriptional target of MESP2 and of TBX6.
- TBX6 encodes a T-box transcription factor. TBX6 activates DLL1 gene expression, which is an activating ligand of the NOTCH1 receptor; it also activates MESP2 gene expression.
Mechanism of disease causation. Loss of function
Table 6.
Autosomal Recessive Spondylocostal Dysostosis: Gene-Specific Laboratory Considerations
Table 7.
Pathogenic Variants Referenced in This GeneReview by Gene
Chapter Notes
Acknowledgments
Research on spondylocostal dysostosis (SCDO) in Exeter has been funded by Action Medical Research, British Scoliosis Research Foundation, and the Skeletal Dysplasia Group, to whom the authors are indebted. In the Exeter Molecular Genetics Laboratory the work was undertaken by Mike Bulman, June Duncan (deceased), Neil Whittock, and recently Melissa Sloman, all under the supervision of Sian Ellard. The work greatly benefited from collaboration with Kenro Kusumi, Sally Dunwoodie, and, more recently, Olivier Pourquié, Philip Giampietro, Alberto Cornier, Amaka Offiah, and Ben Alman, through the ICVAS consortium. Many clinicians have sent images of individuals with segmentation defects of the vertebrae (SDV), but for this review particular thanks are due to Dr Oivind Braaten, Oslo, Norway, and Drs Karin van Spaendonck-Zwarts and Mirjam M de Jong, Groningen, the Netherlands, Professor Eleni Fryysira and Dr Michael Christadoulou, Athens, Greece, and Dr Charles Shaw-Smith, Exeter, UK. Sally Dunwoodie received SCDO research funds from the National Health and Medical Research Council (ID142006, 404804,1044543, 1042002, 1135886).
Author History
Sally Dunwoodie, BSc PhD (2017-present)
Melissa Sloman, BSc, DipRCPath (2017-present)
Peter D Turnpenny, BSc, MB, ChB, FRCP, FRCPCH, FRCPath (2009-present)
Elizabeth Young, PhD; Royal Devon & Exeter NHS Foundation Trust (2009-2017)
Revision History
- 17 August 2023 (sw) Comprehensive update posted live
- 21 December 2017 (sw) Comprehensive update posted live
- 17 January 2013 (me) Comprehensive update posted live
- 25 August 2009 (et) Review posted live
- 6 February 2009 (pdt) Original submission
References
Published Guidelines / Consensus Statements
The ICVAS classification system for congenital scoliosis and segmentation defects of the vertebrae has been published [Turnpenny et al 2007, Offiah et al 2010] and includes an algorithm that helps clinicians determine which individuals are most suitable for genetic testing.
Literature Cited
- Bonafé L, Giunta C, Gassner M, Steinmann B, Superti-Furga A. A cluster of autosomal recessive spondylocostal dysostosis caused by three newly identified DLL3 mutations segregating in a small village. Clin Genet. 2003;64:28-35. [PubMed: 12791036]
- Bonafé L, Superti-Furga A. Martigny, Switzerland: Sixth International Skeletal Dysplasia Society meeting. 2005.
- Bouman A, Waisfisz Q, Admiraal J, van de Loo M, van Rijn RR, Micha D, Oostra R-J, Mathijssen IB. Homozygous DMRT2 variant associates with severe rib malformations in a newborn. Am J Med Genet A. 2018;176:1216-21. [PubMed: 29681102]
- Bulman MP, Kusumi K, Frayling TM, McKeown C, Garrett C, Lander ES, Krumlauf R, Hattersley AT, Ellard S, Turnpenny PD. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat Genet. 2000;24:438-41. [PubMed: 10742114]
- Chen W, Lin J, Wang L, Li X, Zhao S, Liu J, Akdemir ZC, Zhao Y, Du R, Ye Y, Song X, Zhang Y, Yan Z, Yang X, Lin M, Shen J, Wang S, Gao N, Yang Y, Liu Y, Li W, Liu J, Zhang N, Yang X, Xu Y, Zhang J, Delgado MR, Posey JE, Qiu G, Rios JJ, Liu P, Wise CA, Zhang F, Wu Z, Lupski JR, Wu N. TBX6 missense variants expand the mutational spectrum in a non-Mendelian inheritance disease. Human Mutation. 2020;41:182–95. [PMC free article: PMC7061259] [PubMed: 31471994]
- Cornier AS, Ramírez N, Arroyo S, Acevedo J, García L, Carlo S, Korf B. Phenotype characterization and natural history of spondylothoracic dysplasia syndrome: a series of 27 new cases. Am J Med Genet A. 2004;128A:120-6. [PubMed: 15214000]
- Cornier AS, Staehling-Hampton K, Delventhal KM, Saga Y, Caubet JF, Sasaki N, Ellard S, Young E, Ramirez N, Carlo SE, Torres J, Emans JB, Turnpenny PD, Pourquié O. Mutations in the MESP2 gene cause spondylothoracic dysostosis/Jarcho-Levin syndrome. Am J Hum Genet. 2008;82:1334-41. [PMC free article: PMC2427230] [PubMed: 18485326]
- Dequéant ML, Glynn E, Gaudenz K, Wahl M, Chen J, Mushegian A, Pourquié O. A complex oscillating network of signaling genes underlies the mouse segmentation clock. Science. 2006;314:1595-8. [PubMed: 17095659]
- Dequéant ML, Pourquié O. Segmental patterning of the vertebrate embryonic axis. Nat Rev Genet. 2008;9:370-82. [PubMed: 18414404]
- Dolak JA, Tartt S. Spinal anesthesia for Cesarean delivery in a parturient with spondylocostal dysostosis. Can J Anaesth. 2009;56:172-3. [PubMed: 19247767]
- Errichiello E, Arossa A, Iasci A, Villa R, Ischia B, Pavesi MA, Rizzuti T, Bedeschi MF, Zuffardi O. An additional piece in the TBX6 gene dosage model: a novel nonsense variant in a fetus with severe spondylocostal dysostosis. Clin Genet. 2020;98:628–29. [PubMed: 33058178]
- Floor E, De Jong RO, Fryns JP, Smulders C, Vles JS. Spondylocostal dysostosis: an example of autosomal dominant transmission in a large family. Clin Genet. 1989;36:236-41. [PubMed: 2805381]
- Gucev ZS, Tasic V, Pop-Jordanova N, Sparrow DB, Dunwoodie SL, Ellard S, Young E, Turnpenny PD. Autosomal dominant spondylocostal dysostosis in three generations of a Macedonian family: negative mutation analysis of DLL3, MESP2, HES7, and LFNG. Am J Med Genet A. 2010;152A:1378-82. [PubMed: 20503311]
- Jarcho S, Levin PM. Hereditary malformation of the vertebral bodies. Bull Johns Hopkins Hosp. 1938;62:216-26.
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519-22. [PubMed: 28959963]
- Kubryk N, Borde M. La dysostose spondylocostale. Pédiatrie. 1981;17:137-46.
- Lefebvre M, Duffourd Y, Jouan T, Poe C, Jean-Marçais N, Verloes A, St-Onge J, Riviere JB, Petit F, Pierquin G, Demeer B, Callier P, Thauvin-Robinet C, Faivre L, Thevenon J. Autosomal recessive variations of TBX6, from congenital scoliosis to spondylocostal dysostosis. Clin Genet. 2017;91:908-12. [PubMed: 27861764]
- Lefebvre M, Dieux-Coeslier A, Baujat G, Schaefer E, Judith SO, Bazin A, Pinson L, Attie-Bitach T, Baumann C, Fradin M, Pierquin G, Julia S, Quélin C, Doray B, Berg S, Vincent-Delorme C, Lambert L, Bachmann N, Lacombe D, Isidor B, Laurent N, Joelle R, Blanchet P, Odent S, Kervran D, Leporrier N, Abel C, Segers K, Guiliano F, Ginglinger-Fabre E, Selicorni A, Goldenberg A, El Chehadeh S, Francannet C, Demeer B, Duffourd Y, Thauvin-Robinet C, Verloes A, Cormier-Daire V, Riviere JB, Faivre L, Thevenon J. Diagnostic strategy in segmentation defect of the vertebrae: a retrospective study of 73 patients. J. Med Genet. 2018;55:422-9. [PubMed: 29459493]
- Li G, Strong A, Wang H, Kim J-S, Watson D, Zhao S, Vaccaro C, Hartung E, Hakonarson H, Zhang T-J, Giampietro PF, Wu N. TBX6 as a cause of a combined skeletal-kidney dysplasia syndrome. Am J Med Genet A. 2022;188:3469-81. [PMC free article: PMC10473889] [PubMed: 36161696]
- Lorenz P, Rupprecht E. Spondylocostal dysostosis: dominant type. Am J Med Genet. 1990;35:219-21. [PubMed: 2309760]
- McInerney-Leo AM, Sparrow DB, Harris JE, Gardiner BB, Marshall MS, O'Reilly VC, Shi H, Brown MA, Leo PJ, Zankl A, Dunwoodie SL, Duncan EL. Compound heterozygous mutations in RIPPLY2 associated with vertebral segmentation defects. Hum Mol Genet. 2015;24:1234-42. [PubMed: 25343988]
- Offiah A, Alman B, Cornier AS, Giampietro PF, Tassy O, Wade A, Turnpenny PD, et al. Pilot assessment of a radiologic classification system for segmentation defects of the vertebrae. Am J Med Genet A. 2010;152A:1357-71. [PubMed: 20503308]
- Otomo N, Mizumoto S, Lu HF, Takeda K, Campos-Xavier B, Mittaz-Crettol L, Guo L, Takikawa K, Nakamura M, Yamada S, Matsumoto M, Watanabe K, Ikegawa S. Identification of novel LFNG mutations in spondylocostal dysostosis. J Hum Genet. 2019;64:261-4. [PubMed: 30531807]
- Parnell SE, Effmann EL, Song K, Swanson JO, Bompadre V, Phillips GS. Vertical expandable prosthetic titanium rib (VEPTR): a review of indications, normal radiographic appearance and complications. Pediatr Radiol. 2015;45:606-16. [PubMed: 25241040]
- Pons-Odena M, Verges A, Arza N, Cambra FJ. Combined use of neurally adjusted ventilatory assist (NAVA) and vertical expandable prosthetic titanium rib (VEPTR) in a patient with spondylocostal dysostosis and associated bronchomalacia. BMJ Case Rep. 2017;2017:bcr2016217027. [PMC free article: PMC5318610] [PubMed: 28196820]
- Ramirez N, Flynn JM, Emans JB, Betz R, Smith JT, Price N, St Hilaire T, Joshi AP, Campbell RM. Vertical expandable prosthetic titanium rib as treatment of thoracic insufficiency syndrome in spondylocostal dysplasia. J Pediatr Orthop. 2010;30:521-6. [PubMed: 20733413]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rimoin DL, Fletcher BD, McKusick VA. Spondylocostal dysplasia. A dominantly inherited form of short-trunked dwarfism. Am J Med. 1968;45:948-53. [PubMed: 5722643]
- Sandbacka M, Laivuori H, Freitas É, Halttunen M, Jokimaa V, Morin-Papunen L, Rosenberg C, Aittomäki K. TBX6, LHX1 and copy number variations in the complex genetics of müllerian aplasia. Orphanet J Rare Dis. 2013;8:125. [PMC free article: PMC3847609] [PubMed: 23954021]
- Schuhmann S, Koller H, Sticht H, Kraus C, Krumbiegel M, Uebe S, Ekici AB, Reis A, Thiel CT. Clinical and molecular delineation of spondylocostal dysostosis type 3. Clin Genet. 2021;99:851-2. [PubMed: 33728697]
- Serey-Gaut M, Scala M, Reversade B, Ruaud L, Cabrol C, Musacchia F, Torella A, Accogli A, Escande-Beillard N, Langlais J, Piatelli G, Consales A, Nigro V, Capra V, Van Maldergem L. Congenital posterior cervical spine malformation due to biallelic c.240-4T>G RIPPLY2 variant: a discrete entity. Am J Med Genet A. 2020;182:1466-72. [PubMed: 32212228]
- Sparrow DB, Ali Faqeih E, Sallout B, Alswaid A, Ababneh F, Al-Sayed M, Rukban H, Eyaid WM, Kageyama R, Sian Ellard, Turnpenny PD, Dunwoodie SL. Mutation of HES7 in a large extended family with spondylocostal dysostosis and dextrocardia with situs inversus. Am J Med Genet A. 2013a;161A:2244-9. [PubMed: 23897666]
- Sparrow DB, Chapman G, Smith AJ, Mattar MZ, Major JA, O'Reilly VC, Saga Y, Zackai EH, Dormans JP, Alman BA, McGregor L, Kageyama R, Kusumi K, Dunwoodie SL. A mechanism for gene-environment interaction in the etiology of congenital scoliosis. Cell. 2012;149:295-306. [PubMed: 22484060]
- Sparrow DB, Chapman G, Wouters MA, Whittock NV, Ellard S, Fatkin D, Turnpenny PD, Kusumi K, Sillence D, Dunwoodie SL. Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am J Hum Genet. 2006;78:28-37. [PMC free article: PMC1380221] [PubMed: 16385447]
- Sparrow DB, Guillén-Navarro E, Fatkin D, Dunwoodie SL. Mutation of hairy-and-enhancer-of-split-7 in humans causes spondylocostal dysostosis. Hum Mol Genet. 2008;17:3761-6. [PubMed: 18775957]
- Sparrow DB, McInerney-Leo A, Gucev ZS, Gardiner B, Marshall M, Leo PJ, Chapman D L, Tasic V, Shishko A, Brown MA, Duncan EL, Dunwoodie SL. Autosomal dominant spondylocostal dysostosis is caused by mutation in TBX6. Hum Molec Genet. 2013b;22:1625-31. [PubMed: 23335591]
- Sparrow DB, Sillence D, Wouters MA, Turnpenny PD, Dunwoodie SL. Two novel missense mutations in HAIRY-AND-ENHANCER-OF-SPLIT-7 in a family with spondylocostal dysostosis. Eur J Hum Genet. 2010;18:674-9. [PMC free article: PMC2987349] [PubMed: 20087400]
- Studer D, Hasler CC. Long term outcome of vertical expandable prosthetic titanium rib treatment in children with early onset scoliosis. Ann Transl Med. 2020;8:25-31. [PMC free article: PMC6995913] [PubMed: 32055616]
- Takeda K, Kou I, Kawakami N, Iida A, Nakajima M, Ogura Y, Imagawa E, Miyake N, Matsumoto N, Yasuhiko Y, Sudo H, Kotani T, Nakamura M, Matsumoto M, Watanabe K, Ikegawa S, et al. Compound heterozygosity for null mutations and a common hypomorphic risk haplotype in TBX6 causes congenital scoliosis. Hum Mutat. 2017;38:317-23. [PubMed: 28054739]
- Takeda K, Kou I, Mizumoto S, Yamada S, Kawakami N, Nakajima M, Otomo N, Ogura Y, Miyake N, Matsumoto N, Kotani T, Sudo H, Yonezawa I, Uno K, Taneichi H, Watanabe K, Shigematsu H, Sugawara R, Taniguchi Y, Minami S, Nakamura M, Matsumoto M, Watanabe K, Ikegawa S, et al. Screening of known disease genes in congenital scoliosis. Mol Genet Genomic Med. 2018;6:966-74. [PMC free article: PMC6305645] [PubMed: 30196550]
- Temple IK, Thomas TG, Baraitser M. Congenital spinal deformity in a three generation family. J Med Genet. 1988;25:831-4. [PMC free article: PMC1051611] [PubMed: 3236365]
- Turnpenny PD, Alman B, Cornier AS, Giampietro PF, Offiah A, Tassy O, Pourquié O, Kusumi K, Dunwoodie S. Abnormal vertebral segmentation and the notch signaling pathway in man. Dev Dyn. 2007;236:1456-74. [PubMed: 17497699]
- Turnpenny PD, Bulman MP, Frayling TM, Abu-Nasra TK, Garrett C, Hattersley AT, Ellard S. A gene for autosomal recessive spondylocostal dysostosis maps to 19q13.1-q13.3. Am J Hum Genet. 1999;65:175-82. [PMC free article: PMC1378088] [PubMed: 10364530]
- Turnpenny PD, Whittock N, Duncan J, Dunwoodie S, Kusumi K, Ellard S. Novel mutations in DLL3, a somitogenesis gene encoding a ligand for the Notch signalling pathway, cause a consistent pattern of abnormal vertebral segmentation in spondylocostal dysostosis. J Med Genet. 2003;40:333-9. [PMC free article: PMC1735475] [PubMed: 12746394]
- Unger S, Ferreira CR, Mortier GR, Ali H, Bertola DR, Calder A, Cohn DH, Cormier-Daire V, Girisha KM, Hall C, Krakow D, Makitie O, Mundlos S, Nishimura G, Robertson SP, Savarirayan R, Sillence D, Simon M, Sutton VR, Warman ML, Superti-Furga A. Nosology of genetic skeletal disorders: 2023 revision. Am J Med Genet A. 2023;191:1164–209. [PMC free article: PMC10081954] [PubMed: 36779427]
- Urioste M, Lorda-Sánchez I, Blanco M, Burón E, Aparicio P, Martínez-Frías ML. Severe congenital limb deficiencies, vertebral hypersegmentation, absent thymus and mirror polydactyly: a defect expression of a developmental control gene? Hum Genet. 1996;97:214-7. [PubMed: 8566956]
- Waschk DE, Tewes AC, Römer T, Hucke J, Kapczuk K, Schippert C, Hillemanns P, Wieacker P, Ledig S. Mutations in WNT9B are associated with Mayer-Rokitansky-Küster-Hauser syndrome. Clin Genet. 2016;89:590-6. [PubMed: 26610373]
- Whittock NV, Ellard S, Duncan J, de Die-Smulders CE, Vles JS, Turnpenny PD. Pseudodominant inheritance of spondylocostal dysostosis type 1 caused by two familial delta-like 3 mutations. Clin Genet. 2004a;66:67-72. [PubMed: 15200511]
- Whittock NV, Sparrow DB, Wouters MA, Sillence D, Ellard S, Dunwoodie SL, Turnpenny PD. Mutated MESP2 causes spondylocostal dysostosis in humans. Am J Hum Genet. 2004b;74:1249-54. [PMC free article: PMC1182088] [PubMed: 15122512]
- Wu N, Ming X, Xiao J, Wu Z, Chen X, Shinawi M, Shen Y, Yu G, Liu J, Xie H, Gucev ZS, Liu S, Yang N, Al-Kateb H, Chen J, Zhang J, Hauser N, Zhang T, Tasic V, Liu P, Su X, Pan X, Liu C, Wang L, Shen J, Shen J, Chen Y, Zhang T, Zhang J, Choy KW, Wang J, Wang Q, Li S, Zhou W, Guo J, Wang Y, Zhang C, Zhao H, An Y, Zhao Y, Wang J, Liu Z, Zuo Y, Tian Y, Weng X, Sutton VR, Wang H, Ming Y, Kulkarni S, Zhong TP, Giampietro PF, Dunwoodie SL, Cheung SW, Zhang X, Jin L, Lupski JR, Qiu G, Zhang F. TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med. 2015;372:341-50. [PMC free article: PMC4326244] [PubMed: 25564734]
Publication Details
Author Information and Affiliations
University of Exeter Medical School
Royal Devon University Healthcare NHS Foundation Trust
Exeter, United Kingdom
Royal Devon University Healthcare NHS Foundation Trust
Exeter, United Kingdom
Victor Chang Cardiac Research Institute
Sydney, New South Wales, Australia
Faculty of Medicine and Health
University of New South Wales
Sydney, New South Wales, Australia
Publication History
Initial Posting: August 25, 2009; Last Update: August 17, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Turnpenny PD, Sloman M, Dunwoodie S. Spondylocostal Dysostosis, Autosomal Recessive. 2009 Aug 25 [Updated 2023 Aug 17]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.