Summary
Clinical characteristics.
POLR3-related leukodystrophy, a hypomyelinating leukodystrophy with specific features on brain MRI, is characterized by varying combinations of four major clinical findings:
- Neurologic dysfunction, typically predominated by motor dysfunction (progressive cerebellar dysfunction, and to a lesser extent extrapyramidal [i.e., dystonia], pyramidal [i.e., spasticity] and cognitive dysfunctions)
- Abnormal dentition (delayed dentition, hypodontia, oligodontia, and abnormally placed or shaped teeth)
- Endocrine abnormalities such as short stature (in ~50% of individuals) with or without growth hormone deficiency, and more commonly, hypogonadotropic hypogonadism manifesting as delayed, arrested, or absent puberty
- Ocular abnormality in the form of myopia, typically progressing over several years and becoming severe
POLR3-related leukodystrophy and 4H leukodystrophy are the two recognized terms for five previously described overlapping clinical phenotypes (initially described as distinct entities before their molecular basis was known). These include:
- Hypomyelination, hypodontia, hypogonadotropic hypogonadism (4H syndrome);
- Ataxia, delayed dentition, and hypomyelination (ADDH);
- Tremor-ataxia with central hypomyelination (TACH);
- Leukodystrophy with oligodontia (LO);
- Hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum (HCAHC).
Age of onset is typically in early childhood but later-onset cases have also been reported.
An infant with Wiedemann-Rautenstrauch syndrome (neonatal progeroid syndrome) was recently reported to have pathogenic variants in POLR3A on exome sequencing. Confirmation of this as a very severe form of POLR3-related leukodystrophy awaits replication in other individuals with a clinical diagnosis of Wiedemann-Rautenstrauch syndrome.
Diagnosis/testing.
POLR3-related leukodystrophy is diagnosed by the combination of classic clinical findings, typical brain MRI features, and the presence of biallelic pathogenic variants in POLR3A, POLR3B, or POLR1C.
Management.
Treatment of manifestations: Individualized care by a multidisciplinary team including a pediatric neurologist, clinical geneticist, physiotherapist, occupational therapist, speech and language pathologist, neuropsychologist, rehabilitation physician, dentist, endocrinologist, ophthalmologist, ear-nose-and-throat specialist, and primary care physician is recommended. Special caution needs to be taken when managing dysphagia in this disorder as it is known to vary widely, even in a single day. Swallowing difficulties will progress over time and dystonia should be monitored and treated to prevent complications and improve the quality of life.
Genetic counseling.
POLR3-related leukodystrophy is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk relatives and prenatal diagnosis for pregnancies at increased risk are possible if both pathogenic variants in the family are known.
Diagnosis
Suggestive Findings
POLR3-related leukodystrophy should be suspected in individuals with the following major/shared clinical features, which may or may not be present:
- Neurologic dysfunction: progressive cerebellar features, including:
- Gait ataxia, dysarthria, dysmetria, tremor, eye movement abnormalities; and
- To a lesser extent, extrapyramidal (typically dystonia), pyramidal, and cognitive features
- Abnormal dentition (e.g., hypodontia, oligodontia, delayed teeth eruption) [Wolff et al 2010]
- Endocrine abnormalities such as short stature (in ~50% of individuals) with or without growth hormone deficiency, and more commonly, hypogonadotropic hypogonadism manifesting as delayed, arrested, or absent puberty
- Ocular abnormality in the form of myopia, typically progressing over several years and becoming severe
Note: Although this tetrad is highly suggestive of the diagnosis, not all features are present in all individuals who have POLR3-related leukodystrophy.
Brain MRI findings
- A hypomyelinating leukodystrophy pattern characterized by T2 mild hyperintensity of the white matter and T1 hyperintensity, isointensity, or mild hypointensity of the white matter when compared with grey matter structures [Schiffmann & van der Knaap 2009]
- Relative preservation of myelination of specific brain structures, i.e. T2 hypointense signal (normal or almost normal white matter signal) of the dentate nuclei, anterolateral nuclei of the thalami, globi pallidi, pyramidal tracts in the posterior limbs of the internal capsules, and optic radiations [Steenweg et al 2010, La Piana et al 2014, Wolf et al 2014]
- Variably present: cerebellar atrophy and thinning of the corpus callosum [La Piana et al 2014, Wolf et al 2014]
- Rarely, an atypical MRI pattern with either
- Selective hypomyelination of the corticospinal tracts; or
- Cerebellar atrophy with or without focal hypomyelination [La Piana et al 2016]; or
- Involvement of the striata and red nuclei [Azmanov et al 2016]
See Figure 1.
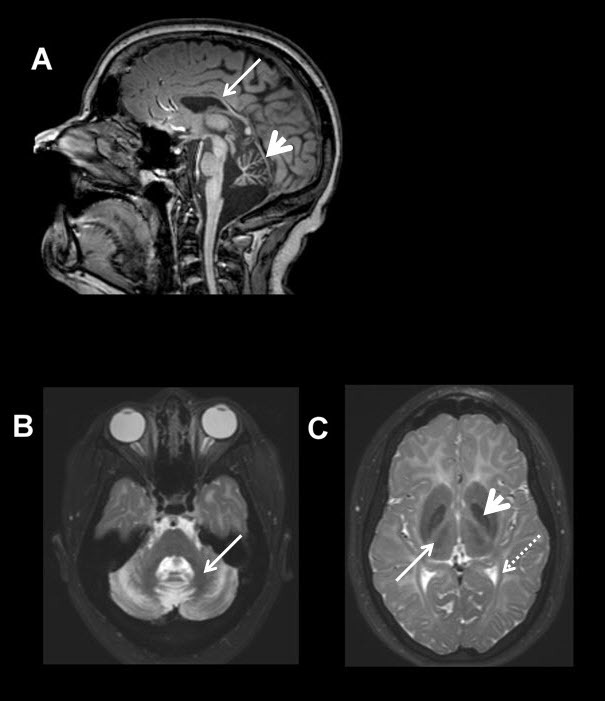
Figure 1.
MRI of the brain of an individual with molecularly confirmed 4H syndrome A. Sagittal T1-weighted image (midline) showing cerebellar atrophy (arrowhead) as well as a thin corpus callosum (arrow).
Establishing the Diagnosis
The diagnosis of POLR3-related leukodystrophy is established in a proband with the combination of classic clinical findings, typical brain MRI features, and identification of biallelic pathogenic (or likely pathogenic) variants in POLR3A, POLR3B, or POLR1C on molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of biallelic variants of uncertain significance (or of one known pathogenic variant and one variant of uncertain significance) in POLR3A, POLR3B, or POLR1C does not establish or rule out the diagnosis.
Molecular testing approaches can include serial single-gene testing, use of a multigene panel, and more comprehensive genomic testing.
Serial single-gene testing. The order of genes to be tested depends on ancestry of the individual:
- In French-Canadian individuals, sequencing of POLR3A should be performed first and followed by gene-targeted deletion/duplication analysis if only one or no pathogenic variant is found.
- If no pathogenic variant is found, sequencing of POLR3B is performed, followed by gene-targeted deletion/duplication analysis if only one or no pathogenic variant is found.
- For individuals of European ancestry sequencing of POLR3B should be performed first.
- If a single variant is detected, gene-targeted deletion/duplication analysis of POLR3B is done.
- If no pathogenic variant is found, sequencing of POLR3A is performed, followed by gene-targeted deletion/duplication analysis if only one or no pathogenic variant is found.
- For individuals from the non-French-Canadian and non-European ancestry, sequencing can start with either POLR3A or POLR3B, followed by gene-targeted deletion/duplication analysis if only one or no pathogenic variant is found.
POLR1C is tested when sequencing and deletion/duplication analysis of POLR3A and POLR3B are both negative.
A multigene panel that includes POLR3A, POLR3B, POLR1C with or without other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included and the sensitivity of multigene panels vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel provides the best opportunity to identify the genetic cause of the condition while limiting identification of pathogenic variants in genes that do not explain the underlying phenotype. (3) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
More comprehensive genomic testing (when available) including exome sequencing and genome sequencing may be considered. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation).
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in POLR3-Related Leukodystrophy
Clinical Characteristics
Clinical Description
POLR3-related leukodystrophy is a hypomyelinating leukodystrophy characterized by neurologic (cerebellar, extrapyramidal, pyramidal, and cognitive) and non-neurologic (dental, endocrine, and ocular) features.
Before the identification of the involved genes, five overlapping clinical phenotypes were described and are now all recognized as part of the spectrum of POLR3-related leukodystrophy:
- 4H syndrome. Hypomyelination, hypodontia, hypogonadotropic hypogonadism [Wolf et al 2005, Timmons et al 2006, Vázquez-López et al 2008]
- ADDH. Ataxia, delayed dentition, and hypomyelination [Wolf et al 2007, Wolff et al 2010]
- TACH. Tremor-ataxia with central hypomyelination [Bernard et al 2010, Bernard et al 2011, Tétreault et al 2011, Tétreault et al 2012]
- LO. Leukodystrophy with oligodontia [Atrouni et al 2003, Chouery et al 2011]
- HCAHC. Hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum [Sasaki et al 2009, Saitsu et al 2011]
Age of onset is typically in early childhood but later-onset cases have also been reported.
Neurologic dysfunction includes prominent cerebellar features leading to progressive gait abnormalities, dysmetria, dysdiadochokinesis, dysarthria, and extraocular movement abnormalities including progressive loss of pursuits (saccadic ocular pursuits), abnormal saccades (i.e., hypo- or hypermetric), and gaze-evoked nystagmus. Progressive vertical more than horizontal gaze restriction has also been reported. Hypersalivation and dysphagia are also commonly seen, typically later in the disease course.
- Individuals may also develop extrapyramidal features – almost always dystonia.
- Mild pyramidal features are typically present but are not problematic in the vast majority of individuals.
- An upper-extremity tremor (usually cerebellar in nature) with or without a dystonic component may be a presenting sign.
- Cognitive decline typically occurs later, except in late-onset cases.
- Approximately 10% of individuals have later onset and slower progression; they typically present with academic difficulties and cognitive decline which manifests as a plateau in learning at school. Those with later onset may have also behavioral abnormalities.
Dental abnormalities include delayed dentition, hypodontia, oligodontia, and eruption of abnormally placed or shaped teeth, among others [Wolff et al 2010]. Dental abnormalities may be subtle and have not been seen in all individuals.
Endocrine abnormalities. Hypogonadotropic hypogonadism (HH), the most typical endocrine characteristic, is note seen in all individuals. When reported, HH typically presents with delayed or arrested puberty or absence of early pubertal changes. Other reported endocrine abnormalities include short stature (in 50% of individuals [Wolf et al 2014]) and growth hormone deficiency [Potic et al 2012, Billington et al 2015, Potic et al 2015].
Ocular abnormality. Myopia, typically progressive over several years and eventually leading to severe myopia, is the most common non-neurologic feature.
Course and life span. The course of POLR3-related leukodystrophy is invariably progressive. Life span depends on the supportive measures put in place to prevent secondary complications. POLR3-related leukodystrophy is considered to be a life-limiting condition; those with earlier onset are at a higher risk for mortality in young adulthood. Individuals with later onset or with a slower progressive disease course could survive later into adulthood (i.e., 4th-5th decade).
Additional features seen less frequently include the following [Wolf et al 2005, Timmons et al 2006, Vázquez-López et al 2008, Bekiesinska-Figatowska et al 2010, Orcesi et al 2010, Wolff et al 2010, Sato et al 2011, Potic et al 2012]:
- Neurologic. Seizures reported in a minority of individuals [Bernard et al 2011, Wolf et al 2014]
- Ocular
- Optic atrophy
- Cataracts reported in four individuals [Sato et al 2011, Wolf et al 2014]
Neurophysiologic investigations. All affected individuals undergoing EMG and nerve conduction studies had normal results [Author, personal observation].
Wiedemann-Rautenstrauch syndrome (neonatal progeroid syndrome). A newborn with features of this disorder (including extreme small size, sparse scalp hair, broad abnormally shaped skull, triangular face with midface retraction, natal teeth, decreased subcutaneous fat, and limb contractures) was recently reported to have biallelic truncating variants in POLR3A on exome sequencing that were predicted to result in little residual POLR3 function. Confirmation of this as a very severe form of POLR3-related leukodystrophy awaits replication in other individuals with a clinical diagnosis of Wiedemann-Rautenstrauch syndrome [Jay et al 2016].
Genotype-Phenotype Correlations
POLR3A
- Individuals with POLR3A pathogenic variants tend to have a later disease onset than those with POLR3B pathogenic variants but more rapid disease progression [Wolf et al 2014].
- A single female infant with Wiedemann-Rautenstrauch syndrome (neonatal progeroid syndrome) was recently reported to have biallelic truncating pathogenic variants in POLR3A [Jay et al 2016].
POLR3B
- Individuals with POLR3B pathogenic variants have earlier disease onset than those with POLR3A pathogenic variants but a slower disease course.
- Those with POLR3B pathogenic variants are more likely to have cerebellar atrophy on MRI and preservation of the myelination of the corticospinal tracts at the level of the internal capsules, compared to individuals with POLR3A pathogenic variants.
- The common POLR3B pathogenic variant c.1568T>A (p.Val523Glu) is very mild. Indeed, individuals homozygous for this variant were either asymptomatic or very mildly symptomatic in early adulthood [Wolf et al 2014].
POLR1C. Limited information is available on individuals with POLR1C pathogenic variants.
Prevalence
The prevalence of POLR3-related leukodystrophy is unknown.
Individuals with POLR3-related leukodystrophy have been identified worldwide [Atrouni et al 2003, Sasaki et al 2009, Bekiesinska-Figatowska et al 2010, Saitsu et al 2011, Sato et al 2011, Wolf et al 2014].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with pathogenic variants in POLR3A or POLR3B.
Different biallelic pathogenic variants POLR1C are associated with Treacher Collins syndrome.
Differential Diagnosis
The differential diagnosis of POLR3-related leukodystrophy includes other hypomyelinating leukodystrophies. See Table 2.
Table 2.
Hypomyelinating Leukodystrophies to Consider in the Differential Diagnosis of POLR3-Related Leukodystrophy
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with a POLR3-related leukodystrophy, the following are recommended:
- Pediatric neurology consultation
- Swallowing assessment
- Physiotherapy evaluation
- Occupational therapy evaluation
- Speech and language pathology assessment
- Rehabilitation physician (i.e., physiatrist) consultation
- Neuropsychology evaluation
- Brain MRI, if not performed at the time of diagnosis
- Dentistry consultation
- Endocrine consultation
- Ophthalmologic evaluation
- Ear-nose-and-throat specialist consultation for hypersalivation and swallowing issues
- Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Individualized care by a multidisciplinary team including a pediatric neurologist, clinical geneticist, physiotherapist, occupational therapist, speech and language pathologist, neuropsychologist, rehabilitation physician, dentist, endocrinologist, ophthalmologist, ear-nose-and-throat specialist, and primary care physician is recommended.
Manifestations such as ambulation difficulties and seizures are managed in a routine manner.
Special caution needs to be taken when managing dysphagia in this disorder as it is known to be quite variable, even in a single day. This is probably due to the prominent cerebellar involvement, leading to more incoordination of swallowing with fatigue, but also with some unpredictability. Dysphagia management is therefore important. Swallowing difficulties will progress over time and individuals may require gastrostomy to manage nutrition.
Spasticity and dystonia should be monitored and treated to prevent complications and to improve the quality of life.
Hypersalivation is managed with the multidisciplinary team and an otolaryngologist. Treatment must be individualized. Therapies to consider include rehabilitation (e.g., oromotor therapy, behavioral therapy); medical therapy (e.g., anticholinergic medications, botulinum toxin injections); and, in severe cases, surgery (e.g., relocation of the parotid ducts and relocation of the submandibular ducts with or without sublingual gland excision).
Learning difficulties are likely to progress slowly. The cognitive involvement is typically much less severe than the motor involvement. In contrast, activities of daily living are likely to become problematic early on in the disease course as motor difficulties make individuals progressively more dependent on assistance from others.
Dental manifestations should be managed, when necessary, by a dentist and/or orthodontist.
Affected individuals should be followed regularly by an endocrinologist. The decision to treat sex and growth hormone deficiency, when present, should be made on an individual basis.
Individuals need to be followed regularly by an ophthalmologist. A significant proportion of affected individuals will have progressive myopia over several years, which will become severe before stabilizing.
Surveillance
No general surveillance guidelines have been developed to date; monitoring should be individualized.
Agents/Circumstances to Avoid
Avoid the following:
- Foods that are likely to lead to choking
- Medications acting on D2 receptor blockers (e.g., neuroleptics such as haloperidol or risperidone, anti-nausea medications such as metoclopramide) as these can exacerbate the extrapyramidal features
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
POLR3-related leukodystrophy is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., carriers of one POLR3A, POLR3B, or POLR1C pathogenic variant).
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Note: The phenotype is similar among family members; however, the severity can vary.
- Heterozygotes (carriers) are asymptomatic.
Offspring of a proband. The offspring of an individual with POLR3-related leukodystrophy are obligate heterozygotes (i.e., carriers of one POLR3A, POLR3B, or POLR1C pathogenic variant).
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a POLR3A, POLR3B, or POLR1C pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the POLR3A, POLR3B, or POLR1C pathogenic variants in the family.
Related Genetic Counseling Issues
Predictive testing for at-risk asymptomatic adult family members requires prior identification of the pathogenic variants in the family. Note: Although the phenotype is similar among family members, the severity and age of onset can vary.
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are carriers or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Once the POLR3A, POLR3B, or POLR1C pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing for POLR3-related leukodystrophy are possible.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- The Yaya Foundation for 4H LeukodystrophyPO Box 80685Minneapolis MN 55408Email: info@yayafoundation4HL.org
- European Leukodystrophy Association (ELA)Phone: 03 83 30 93 34
- Leukodystrophy AustraliaAustraliaPhone: 1800-141-400Email: info@leuko.org.au
- Leukodystrophy Foundation1713, rue des Épinettes-RougesQuébec City Quebec G3G 2L2CanadaPhone: 418 806-2968Fax: 1 866 421-6846Email: info@leucofondation.com
- Medical Home Portal
- United Leukodystrophy FoundationPhone: 800-SAV-LIVE; 815-748-3211Email: office@ulf.org
- Myelin Disorders Bioregistry ProjectPhone: 215-590-1719Email: sherbinio@chop.edu
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
POLR3-Related Leukodystrophy: Genes and Databases
Table B.
OMIM Entries for POLR3-Related Leukodystrophy (View All in OMIM)
Molecular Pathogenesis
Three types of RNA polymerases (Pol I [POLR1], Pol II [POLR2], and Pol III [POLR3]) are responsible for the transcription of DNA into RNA [Cramer et al 2008, Werner et al 2009]. POLR1, POLR2, and POLR3 have a distinct repertoire of DNA targets. POLR3A and POLR3B are the two largest subunits of POLR3 and form the catalytic core of the polymerase III. POLR1C is a shared subunit to POLR1 and POLR3.
- POLR1 transcribes pre-rRNAs, which are modified into most of the ribosomal RNAs constituting the 18S, 5.8S, and 28S ribosomal subunits.
- POLR2 transcribes all protein-coding genes and multiple noncoding genes, including the majority of microRNAs (miRNAs).
- POLR3 has a diverse repertoire of nuclear targets and also transcribes exogenous DNA in the cytoplasm.
- In the nucleus it transcribes all tRNAs (transfer RNAs), the RNA 7SL (which is necessary for the insertion of proteins into membranes), RNAs 7SK, Alu, and B2 elements (which are responsible for the regulation of the transcription of POLR2).
- POLR3 may play a role in the transcription of some miRNAs, but not to the same extent as POLR2 [Dieci et al 2007].
- POLR3 is responsible for the transcription of the ribosomal subunit 5S gene, which is involved in cytoplasmic and mitochondrial translation [Szymański et al 2003].
- POLR3 transcribes vault RNAs, which become the vault organelles (responsible for the transport of the mRNAs from the nucleus to the cytoplasm).
POLR3A
Gene structure. POLR3A comprises 31 exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. Pathogenic variants include missense, nonsense, and splice site variants, small intragenic deletions, and insertions [Bernard et al 2011, Saitsu et al 2011, Potic et al 2012, Daoud et al 2013, Wolf et al 2014, La Piana et al 2016]. See Table 3 (pdf).
No affected individual has been found to have two null pathogenic variants, which may be explained by the role of POLR3A as a housekeeping gene, suggesting that some residual POLR3 function is essential for embryonic/fetal survival.
Normal gene product. POLR3A encodes the DNA-directed RNA polymerase III subunit A (POLR3A) which comprises 1390 amino acids. POLR3A, the largest of the 17 subunits of POLR3, forms (together with POLR3B) the catalytic core of the polymerase.
Abnormal gene product. The mapping of the pathogenic variants to protein domains of POLR3A suggests that they can interfere with DNA binding directly, modify the catalytic cleft structure, change POLR3A/POLR3B interaction, and perturb interactions between POLR3A and other POLR3 subunits [Bernard et al 2011].
Western blot studies on fibroblasts and brain tissue from an individual with 4H leukodystrophy and known POLR3A pathogenic variants showed a statistically significant decrease in POLR3A levels, with a more significant reduction in the cerebral white matter compared to the cortex [Bernard et al 2011].
These later findings have led us to hypothesize that pathogenic variants in POLR3A lead to suboptimal assembly of the POLR3 complex, its translocation in the nucleus and/or interaction with chromatin. This hypothesis is supported by the results published on POLR1C leukodystrophy pathogenic variants.
POLR3B
Gene structure. POLR3B comprises 28 exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. Pathogenic variants include missense, nonsense, and splice site variants and small intragenic deletions [Saitsu et al 2011, Tétreault et al 2011]. See Table 4 (pdf). Recently, the first examples of large multiexon deletions have been reported [Gutierrez et al 2015].
No affected individual has been found to have two null pathogenic variants, which may be explained by the role of POLR3B as a housekeeping gene, suggesting that some residual POLR3 function is essential for embryonic/fetal survival.
Normal gene product. POLR3B encodes the DNA-directed RNA polymerase III subunit B (POLR3B) which comprises 1133 amino acids. POLR3B, the second largest of the 17 subunits of POLR3, forms (together with POLR3A) the catalytic core of the polymerase.
Abnormal gene product. The mapping of the pathogenic variants to protein domains of POLR3B suggests that they can interfere with DNA binding directly, modify the catalytic cleft structure, change POLR3A/POLR3B interaction, and perturb interactions between POLR3B and other POLR3 subunits [Tétreault et al 2011].
POLR1C
Gene structure. POLR1C comprises nine exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. Pathogenic leukodystrophy variants reported to date include missense and small intragenic deletions different from the pathogenic variants known to cause Treacher Collins syndrome. See Table 5 (pdf).
Normal gene product. POLR1C encodes the DNA-directed RNA polymerase I subunit C (POLR1C) which comprises 346 amino acids. POLR1C is a common subunit to both POLR1 and POLR3.
Abnormal gene product. The abnormal "leukodystrophy" gene products p.Asn32Ile and p.Asn74Ser alter assembly and nuclear import of POLR3, but not POLR1, leading to decreased binding to POLR3 target genes, and not POLR1 target genes [Thiffault et al 2015].
Chapter Notes
Author Notes
Contact information for Dr Geneviève Bernard's laboratory at the Research Institute of the McGill University Health Centre:
Pediatric Neurodegenerative Laboratory
1001 boul Décarie
Site Glen Pavilion E / Block E
CHHD Mail Drop Point #EM03211 (cubicle C )
Montréal, QC H4A 3J1
Canada
Email: genevieve.bernard@mcgill.ca
Phone: 514-934-1934 ext 23380
Acknowledgments
Drs Bernard and Vanderver wish to thank all patients and their families as well as all the collaborators involved in the POLR3-related leukodystrophy project. Dr Bernard has received a Research Scholar Junior 1 award from the Fonds de Recherche du Québec en Santé (FRQS) 2012-2016 and a Canadian Institute of Health Research New Investigator salary award (2017-2022) (201512MSH-360766-171036). She wishes to thank the Canadian Institutes of Health Research (CIHR MOP-G-287547 and MOP-G2-341146-159133-BRIDG), the Réseau de Médecine Génétique Appliquée (RMGA), the Leuko Dystrophy Foundation, the "Fondation du Grand Défi Pierre Lavoie," the European Leukodystrophy Association, and the "Fondation les Amis D'Elliot," for financing her research projects on leukodystrophies. Dr Vanderver wishes to thank the Intramural Research Program of the National Human Genome Research Institute and the Myelin Disorders Bioregistry Project.
Revision History
- 11 May 2017 (ha) Comprehensive update posted live
- 2 August 2012 (me) Review posted live
- 1 March 2012 (av) Original submission
References
Published Guidelines / Consensus Statements
- Van Haren K, Bonkowsky JL, Bernard G, Murphy JL, Pizzino A, Helman G, Suhr D, Waggoner J, Hobson D, Vanderver A, Patterson MC, et al. Consensus statement on preventive and symptomatic care of leukodystrophy patients. Mol Genet Metab. 2015;114:516-26. [PubMed]
Literature Cited
- Atrouni S, Darazé A, Tamraz J, Cassia A, Caillaud C, Mégarbané A. Leukodystrophy associated with oligodontia in a large inbred family: fortuitous association or new entity? Am J Med Genet A. 2003;118A:76-81. [PubMed: 12605447]
- Azmanov DN, Siira SJ, Chamova T, Kaprelyan A, Guergueltcheva V, Shearwood AJ, Liu G, Morar B, Rackham O, Bynevelt M, Grudkova M, Kamenov Z, Svechtarov V, Tournev I, Kalaydjieva L, Filipovska A. Transcriptome-wide effects of a POLR3A gene mutation in patients with an unusual phenotype of striatal involvement. Hum Mol Genet. 2016;25:4302-14. [PubMed: 27506977]
- Bekiesinska-Figatowska M, Mierzewska H, Kuczynska-Zardzewialy A, Szczepanik E, Obersztyn E. Hypomyelination, hypogonadotropic hypogonadism, hypodontia - First Polish patient. Brain Dev. 2010;32:574-8. [PubMed: 19700253]
- Bernard G, Chouery E, Putorti ML, Tétreault M, Takanohashi A, Carosso G, Clément I, Boespflug-Tanguy O, Rodriguez D, Delague V, Abou Ghoch J, Jalkh N, Dorboz I, Fribourg S, Teichmann M, Megarbane A, Schiffmann R, Vanderver A, Brais B. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89:415-23. [PMC free article: PMC3169829] [PubMed: 21855841]
- Bernard G, Thiffault I, Tétreault M, Putorti ML, Bouchard I, Sylvain M, Melancon S, Laframboise R, Langevin P, Bouchard JP, Vanasse M, Vanderver A, Sebire G, Brais B. Tremor-ataxia with central hypomyelination (TACH) leukodystrophy maps to chromosome 10q22.3-10q23.31. Neurogenetics. 2010;11:457-64. [PMC free article: PMC4147760] [PubMed: 20640464]
- Billington E, Bernard G, Gibson W, Corenblum B. Endrocrine aspects of 4H leukodystrophy: a case report and review of the literature. Case Rep Endocrinol. 2015;2015:314594. [PMC free article: PMC4465690] [PubMed: 26113998]
- Chouery E, Delague V, Jalkh N, Salem N, Kfoury J, Rodriguez D, Chabrol B, Boespflug-Tanguy O, Lévy N, Serre JL, Mégarbané A. A whole-genome scan in a large family with leukodystrophy and oligodontia reveals linkage to 10q22. Neurogenetics. 2011;12:73-8. [PubMed: 20721593]
- Cramer P, Armache KJ, Baumli S, Benkert S, Brueckner F, Buchen C, Damsma GE, Dengl S, Geiger SR, Jasiak AJ, Jawhari A, Jennebach S, Kamenski T, Kettenberger H, Kuhn CD, Lehmann E, Leike K, Sydow JF, Vannini A. Structure of eukaryotic RNA polymerases. Annu Rev Biophys. 2008;37:337-52. [PubMed: 18573085]
- Daoud H, Tétreault M, Gibson W, Guerrero K, Cohen A, Gburek-Augustat J, Synofzik M, Brais B, Stevens CA, Sanchez-Carpintero R, Goizet C, Naidu S, Vanderver A, Bernard G. Mutations in POLR3A and POLR3B are a major cause of hypomyelinating leukodystrophies with or without dental abnormalities and/or hypogonadotropic hypogonadism. J Med Genet. 2013;50:194-7. [PubMed: 23355746]
- Dieci G, Fiorino G, Castelnuovo M, Teichmann M, Pagano A. The expanding RNA polymerase III transcriptome. Trends Genet. 2007;23:614-22. [PubMed: 17977614]
- Gutierrez M, Thiffault I, Guerrero K, Martos-Moreno G, Tran L, Benko W, van der Knaap M, van Spaendonk R, Wolf N, Bernard G. Large exonic deletions in POLR3B gene cause POLR3-related leukodystrophy. Orphanet J Rare Dis. 2015;10:69. [PMC free article: PMC4520020] [PubMed: 26045207]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389-97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Jay AM, Conway RL, Thiffault I, Saunders C, Farrow E, Adams J, Toriello HV. Neonatal progeriod syndrome associated with biallelic truncating variants in POLR3A. Am J Med Genet A. 2016;170:3343-6. [PubMed: 27612211]
- Jurkiewicz E, Dunin-Wasowicz D, Gieruszczak-Bialek D, Malczyk K, Guerrero K, Gutierrez M, Tran L, Bernard G. Recessive mutations in POLR3B encoding RNA polymerase III subunit causing diffuse hypomyelination in patients with 4H leukodystrophy with polymicrogyria and cataracts. Clin Neuroradiol. 2017;27:213-20. [PMC free article: PMC5487884] [PubMed: 26478204]
- La Piana R, Cayami FK, Tran LT, Guerrero K, van Spaendonk R, Ounap K, Pajusalu S, Haack T, Wassmer E, Timmann D, Mierzewska H, Poll-The BT, Patel C, Cox H, Atik T, Onay H, Ozkinay F, Vanderver A, van der Knaap MS, Wolf NI, Bernard G. Diffuse hypomyelination is not obligate for POLR3-related disorders. Neurology. 2016;86:1622-6. [PMC free article: PMC4844237] [PubMed: 27029625]
- La Piana R, Tonduti D, Gordish Dressman H, Schmidt JL, Murnick J, Brais B, Bernard G, Vanderver A. Brain magnetic resonance imaging (MRI) pattern recognition in Pol III-related leukodystrophies. J Child Neurol. 2014;29:214-20. [PubMed: 24105487]
- Orcesi S, Tonduti D, Uggetti C, Larizza D, Fazzi E, Balottin U. New case of 4H syndrome and a review of the literature. Pediatr Neurol. 2010;42:359-64. [PubMed: 20399393]
- Potic A, Brais B, Choquet K, Schiffmann R, Bernard G. 4H syndrome with late-onset growth hormone deficiency caused by POLR3A mutations. Arch Neurol. 2012;69:920-3. [PubMed: 22451160]
- Potic A, Popovic V, Ostojic J, Pekic S, Kozic D, Guerrero K, Schiffmann R, Bernard G. Neurogenic bladder and neuroendocrine abnormalities in Pol III-related leukodystrophy. BMC Neurol. 2015;15:22. [PMC free article: PMC4351912] [PubMed: 25868523]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Saitsu H, Osaka H, Sasaki M, Takanashi JI, Hamada K, Yamashita A, Shibayama H, Shiina M, Kondo Y, Nishiyama K, Tsurusaki Y, Miyake N, Doi H, Ogata K, Inoue K, Matsumoto N. Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am J Hum Genet. 2011;89:644-51. [PMC free article: PMC3213392] [PubMed: 22036171]
- Sasaki M, Takanashi J, Tada H, Sakuma H, Furushima W, Sato N. Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum. Brain Dev. 2009;31:582-7. [PubMed: 18851904]
- Sato I, Onuma A, Goto N, Sakai F, Fujiwara I, Uematsu M, Osaka H, Okahashi S, Nonaka I, Tanaka S, Haginoya K. A case with central and peripheral hypomyelination with hypogonadotropic hypogonadism and hypodontia (4H syndrome) plus cataract. J Neurol Sci. 2011;300:179-81. [PubMed: 20884016]
- Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72:750-9. [PMC free article: PMC2677542] [PubMed: 19237705]
- Shimojima K, Shimada S, Tamasaki A, Akaboshi S, Komoike Y, Saito A, Furukawa T, Yamamoto T. Novel compound heterozygous mutations of POLR3A revealed by whole-exome sequencing in a patient with hypomyelination. Brain Dev. 2014;36:315-21. [PubMed: 23694757]
- Steenweg ME, Vanderver A, Blaser S, Bizzi A, de Koning TJ, Mancini GM, van Wieringen WN, Barkhof F, Wolf NI, van der Knaap MS. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain. 2010;133:2971-82. [PMC free article: PMC3589901] [PubMed: 20881161]
- Szymański M, Barciszewska MZ, Erdmann VA, Barciszewski J. 5 S rRNA: structure and interactions. Biochem J. 2003;371:641-51. [PMC free article: PMC1223345] [PubMed: 12564956]
- Takanashi J, Osaka H, Saitsu H, Sasaki M, Mori H, Shibayama H, Tanaka M, Nomura Y, Terao Y, Inoue K, Matsumoto N, Barkovich AJ. Different patterns of cerebellar abnormality and hypomyelination between POLR3A and POLR3B mutations. Brain Dev. 2014;36:259-63. [PubMed: 23643445]
- Tétreault M, Choquet K, Orcesi S, Tonduti D, Balottin U, Teichmann M, Fribourg S, Schiffmann R, Brais B, Vanderver A, Bernard G. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89:652-5. [PMC free article: PMC3213403] [PubMed: 22036172]
- Tétreault M, Putorti ML, Thiffault I, Sylvain M, Venderver A, Schiffmann R, Brais B, Bernard G. TACH leukodystrophy: locus refinement to chromosome 10q22.3-23.1. Can J Neurol Sci. 2012;39:122-3. [PubMed: 22384513]
- Thiffault I, Wolf NI, Forget D, Guerrero K, Tran LT, Choquet K, Lavallee-Adam M, Poitras C, Brais B, Yoon G, Sztriha L, Webster RI, Timmann D, van de Warrenburg BP, Seeger J, Zimmermann A, Mate A, Goizet C, Fung E, van der Knaap MS, Fribourg S, Vanderver A, Simons C, Taft RJ, Yates III JR, Coulombe B, Bernard G. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun. 2015;6:7623-31. [PMC free article: PMC4506509] [PubMed: 26151409]
- Timmons M, Tsokos M, Asab MA, Seminara SB, Zirzow GC, Kaneski CR, Heiss JD, van der Knaap MS, Vanier MT, Schiffmann R, Wong K. Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia. Neurology. 2006;67:2066-9. [PMC free article: PMC1950601] [PubMed: 17159124]
- Vázquez-López M, Ruiz-Martín Y, de Castro-Castro P, Garzo-Fernández C, Martín-del Valle F, Márquez-de la Plata L. [Central hypomyelination, hypogonadotrophic hypogonadism and hypodontia: a new leukodystrophy]. Rev Neurol. 2008;47:204-8. [PubMed: 18671210]
- Werner M, Thuriaux P, Soutourina J. Structure-function analysis of RNA polymerases I and III. Curr Opin Struct Biol. 2009;19:740-5. [PubMed: 19896367]
- Wolf NI, Harting I, Boltshauser E, Wiegand G, Koch MJ, Schmitt-Mechelke T, Martin E, Zschocke J, Uhlenberg B, Hoffmann GF, Weber L, Ebinger F, Rating D. Leukoencephalopathy with ataxia, hypodontia, and hypomyelination. Neurology. 2005;64:1461-4. [PubMed: 15851747]
- Wolf NI, Harting I, Innes AM, Patzer S, Zeitler P, Schneider A, Wolff A, Baier K, Zschocke J, Ebinger F, Boltshauser E, Rating D. Ataxia, delayed dentition and hypomyelination: a novel leukoencephalopathy. Neuropediatrics. 2007;38:64-70. [PubMed: 17712733]
- Wolf NI, Vanderver A, van Spaendonk RM, Schiffmann R, Brais B, Bugiani M, Sistermans E, Catsman-Berrevoets C, Kros JM, Pinto PS, Pohl D, Tirupathi S, Stromme P, de Grauw T, Fribourg S, Demos M, Pizzino A, Naidu S, Guerrero K, van der Knaap MS, Bernard G, 4H Research Group. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology. 2014;83:1898-905 [PMC free article: PMC4248461] [PubMed: 25339210]
- Wolff A, Koch MJ, Benzinger S, van Waes H, Wolf NI, Boltshauser E, Luder HU. Rare dental peculiarities associated with the hypomyelinating leukoencephalopathy 4H syndrome/ADDH. Pediatr Dent. 2010;32:386-92. [PubMed: 21070704]
Publication Details
Author Information and Affiliations
Department of Medical Genetics, McGill University Health Center
Child Health and Human Development Program, Research Institute of the McGill University Health Center
Montreal, Quebec, Canada
Children's Research Institute
Children's Hospital of Philadelphia
Philadelphia, Pennsylvania
Publication History
Initial Posting: August 2, 2012; Last Update: May 11, 2017.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Bernard G, Vanderver A. POLR3-Related Leukodystrophy. 2012 Aug 2 [Updated 2017 May 11]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.