

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Auerbach S, Casey W, Chang D, et al. Scientific Studies Supporting Development of Transcriptomic Points of Departure for EPA Transcriptomic Assessment Products (ETAPs). Washington (DC): U.S. Environmental Protection Agency; 2024 Mar.

Scientific Studies Supporting Development of Transcriptomic Points of Departure for EPA Transcriptomic Assessment Products (ETAPs).
Show details4.1. OVERVIEW OF THE APPROACH TO DERIVE TRANSCRIPTOMIC POINTS OF DEPARTURE FOR ETAP
The approach used to derive the transcriptomic PODs for ETAP is largely based on the methodology outlined in the peer-reviewed report entitled National Toxicology Program Approach to Genomic Dose Response Modeling (NTP 2018). The NTP approach outlined in that report originated from a series of publications that developed and refined study designs for quantitative transcriptomic evaluation for dose response assessment and adapted dose response modeling methods used by the EPA for apical endpoints to transcriptomic data (Black et al. 2014; Rowlands et al. 2013; Thomas et al. 2007; Thomas et al. 2011; Thomas et al. 2012; Thomas et al. 2013b). The study design outlined in the NTP approach and used in the ETAP is a 5-day, repeated dose in vivo study in male and female rats with an extended dose response range at multiple dose levels. In the original NTP studies, transcriptomic measurements were performed on the liver and kidneys as sentinel tissues using the BioSpyder TempO-Seq rat S1500+ platform. However, for the 5-day, repeated dose studies for ETAP, transcriptional measurements will be performed on a larger number of tissues to increase the breadth of biological responses evaluated. The tissues will include kidney, liver, adrenal gland, brain, heart, lung, ovary (females), spleen, testis (males), thyroid, thymus, and uterus (females). The TempO-Seq rat S1500+ platform will be used in the ETAP as a pragmatic choice that provides a balance between a set of curated genes that can be cost-effectively employed across multiple tissues, doses, and chemicals and the need to cover important toxicological and disease processes (Mav et al. 2018).
The transcriptomic dose response modeling approach employed in the ETAP is aligned with the NTP report and follows four steps: 1) pre-modeling dataset evaluation to determine adequate signal; 2) pre-modeling probe filtering to remove those that are not responding to treatment; 3) dose response modeling of the individual probes, identifying the best-fit model, and deriving BMD(L) values; and 4) combining the individual probes into gene sets and summarizing the transcriptional BMD(L) values. Consistent with the NTP approach, the transcriptional dose response results from the gene set with the lowest median BMD value will be used to derive the POD for the ETAP (NTP 2018). Transcriptomic BMD values from the gene set with the lowest median BMD value following 5 days of exposure have been demonstrated to be concordant with non-cancer and cancer phenotypic responses in subchronic and chronic toxicity studies (see review in Section 3). The coordinated transcriptional changes used to identify the POD do not necessarily discriminate between specific hazards, adaptive or adverse effects, nor are they used to infer a mechanism or mode of action. Rather, the transcriptomic POD is used to define the experimentally determined dose at which there were no coordinated transcriptional changes that could indicate a potential toxicity of concern.
4.2. ANALYSIS TO SELECT STUDY DESIGN AND TRANSCRIPTOMIC PLATFORM SPECIFIC DOSE RESPONSE MODELING PARAMETERS
The NTP Approach to Genomic Dose Response Modeling report provided general recommendations for the selection of settings and parameters for each step in the dose response modeling process; however, some of the recommendations were acknowledged to be platform specific or provided with minimal data supporting them. The NTP report suggested evaluating various settings and parameter choices to identify an optimal combination that increases detection of true signal, minimizes false signal, and maximizes reproducibility (NTP 2018). To address this suggestion, a comprehensive analysis was undertaken to identify and support the choices and parameters used in each step of the transcriptomic dose response modeling process. Subsets of the data from two NTP datasets were specifically used to address the three goals outlined in the NTP approach (Fig. 4-1; orange boxes). From the first NTP dataset (Gwinn et al. 2020), transcriptomic dose response data for 14 chemicals with data from chronic rodent bioassays were used to evaluate settings and parameters for each component of the dose response modeling process with respect to dose concordance of transcriptional and apical responses (i.e., increase detection of true signal). From the second NTP dataset26, transcriptomic data for three chemicals each with multiple independent replicates were used to evaluate settings and parameters for each component of the dose response modeling process with respect to inter-study reproducibility (i.e., maximize reproducibility). Lastly, combined vehicle control data from both studies were used to evaluate settings and parameters for each component of the dose response modeling process with respect to the family-wise error rate (i.e., minimizing false signal). The two publications used the 5-day, repeated dose in vivo rat study design and BioSpyder TempO-Seq rat S1500+ platform. Therefore, conclusions regarding optimal settings and parameter choices are directly applicable to the ETAP studies.
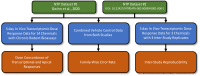
Figure 4-1
Overview of the analysis performed to select study design and transcriptomic platform specific dose response modeling parameters. Subsets of data from two NTP datasets (Gwinn et al. 2020; https://doi.org/10.22427/NTP-DATA-002-00099-0001-000-1) were used (more...)
4.2.1. DOSE CONCORDANCE OF TRANSCRIPTIONAL AND APICAL RESPONSES
4.2.1.1. Overview
To evaluate which dose response modeling parameters provided the best dose concordance between transcriptional and apical responses, the transcriptomic data for the individual chemicals were analyzed using 48 different combinations of selected pre-modeling probe filtering, dose response modeling, and gene set summarization parameters. For each parameter combination, the transcriptomic BMD based on the gene set with the lowest median BMD value was then compared with the minimum of the non-cancer and cancer apical BMD values from the chronic toxicity study. The combinations of parameters were rank ordered based on the RMSD. The parameter combination with the lowest RMSD was selected as the optimal combination for use in the ETAP.
4.2.1.2. Identification of Chronic Apical BMD Values
In the original publication, a subset of 17 out of 19 chemicals had corresponding two-year chronic rodent bioassays in the male rat (Gwinn et al. 2020). Three of these substances, tetrabromobisphenol A, ginseng, and milk thistle extract, did not result in statistically significant apical effects in male rats after two-years of exposure. Since publication of the study, histopathological results from a two-year chronic rodent bioassay for tris(2-chloroisopropyl) phosphate were released,27 and in 2021 a NTP technical report for a two-year chronic rodent bioassay on di(2-ethylhexyl) phthalate was published (NTP 2021). The Gwinn et al. study relied on an earlier NTP technical report for di(2-ethylhexyl) phthalate that was published in 1982. The total number of chemicals from Gwinn et al. study with a two-year chronic rodent bioassay in the male rat in which a statistically significant apical effect was reported is 14. The minimum of the non-cancer and cancer apical BMD values from the chronic study are provided in Table 4-1. Details on the calculation of the apical BMD values are provided in the Appendix (Section 6.1).
Table 4-1
Adverse responses based on the minimum BMD values among non-cancer and cancer histopathological endpoints for 14 chemicals with chronic two-year rodent bioassays.
4.2.1.3. Transcriptomic BMD Modeling Calculations
The transcriptomic dose response modeling process can be broken down into four main steps: 1) pre-modeling dataset evaluation to determine adequate signal; 2) pre-modeling probe filtering to remove those that are not responding to treatment; 3) dose response modeling of the individual probes, identifying the best-fit model, and deriving BMD(L) values; and 4) combining the individual probes into gene sets and summarizing the transcriptional BMD(L) values. At each step in the process, different choices and parameter values are employed for a variety of statistical, biological, and practical reasons including ensuring adequate fit of the dose response model; removing noisy genes or probes; and ensuring sufficient transcriptional responses at the gene set level. As noted in the NTP report, some of the settings and parameter values were acknowledged to be study design or transcriptomic platform specific, while others are not typically study design or platform dependent.
The raw sequencing reads (FASTQ files) for the 14 chemicals with adverse apical outcomes in chronic rodent bioassays were obtained from the NTP. The methods for aligning, normalizing, and quality control of the sequencing data are outlined in the Appendix (Section 6.2). Following quality control, a pre-modeling dataset evaluation was performed on each treatment group using an analysis of variance (ANOVA) with a cut-off of at least one probe showing statistical significance at a Benjamini and Hochberg False Discovery Rate (FDR) corrected p-value < 0.05 (NTP 2018)(See Section 4.2.3 for evaluation of this step). If the treatment group passed the pre-modeling evaluation, transcriptomic dose response modeling was performed. In the transcriptomic dose response modeling, selected BMD modeling settings and parameters were varied to evaluate the impact on the dose concordance between transcriptomic and apical responses. A total of 48 different settings and parameter combinations were evaluated.
For the pre-modeling probe filtering step, the NTP genomics report recommended a combination of statistical significance based on a William’s Trend test combined with a minimum effect size (i.e., fold-change relative to control)(NTP 2018). However, the values associated with these settings may be study design or transcriptomic platform specific. In this analysis, William’s Trend test p-value cut-offs of 0.05 and 0.1 were evaluated together with minimum absolute fold-change cut-offs of 1.5- and 2.0-fold (Table 4-2).
Table 4-2
Transcriptomic BMD modeling settings and parameters that were fixed or varied in the dose concordance analysis.
For the BMD modeling, many of the choices and parameters were fixed given they are not inherently study design or transcriptomic platform dependent. Model fitting was performed on each probe. Linear, second-degree polynomial, power, Hill, second degree exponential, third degree exponential, fourth degree exponential, and fifth degree exponential models were fit to the dose response curves assuming constant variance. The exponent for the power model was restricted (>=1). The model with the lowest Akaike information criterion (AIC) was selected as the best-fit model except in cases where the “k” parameter for the Hill model is less than one-third the lowest dose. In these cases, where the “k” parameter for the Hill model was out of bounds, the Hill model was excluded from the final selection (Rowlands et al. 2013; Thomas et al. 2013b). The Benchmark Response (BMR) was set to 1.349 * standard deviation of replicate vehicle control samples (Thomas et al. 2007). The BMR is different from the recommendation in the NTP genomics report, which listed 1 standard deviation (NTP 2018). Based on EPA guidance, the 1 standard deviation for continuous data is equivalent to a 10% increase in risk for normally distributed effects when the direction of the effects is known (EPA 2012). However, for most gene expression changes, the direction is not known a priori. To provide an equivalent 10% increase in risk, a BMR of 1.349 * standard deviation is required (Thomas et al. 2007). Probes with a BMD greater than the highest dose or a goodness-of-fit p-value less than 0.1 were removed from the analysis. Apart from the fixed BMD modeling parameters, the NTP genomics report recommended removing probes with a high uncertainty in the BMD by applying a BMDU/BMDL filter >40 (NTP 2018). However, the noise associated with BMD values may be study design and transcriptomic platform specific. As a result, two different BMD uncertainty filters (BMD/BMDL>20 and BMDU/BMDL>40) were evaluated (Table 4-2).
For gene set summarization, the GO biological processes were among the gene sets recommended in the NTP approach (NTP 2018). The median BMD and BMDL values were also recommended for summarizing the gene set level potencies (NTP 2018). However, the minimum number of genes in the gene set and the minimum gene set coverage may be transcriptomic platform dependent, especially given the measurement of a smaller number of genes using the S1500+ assay. In this analysis, different cut-offs for the minimum number of genes (3 and 5) and minimum gene set coverage (0%, 3%, and 5%) were evaluated (Table 4-2).
4.2.1.4. Evaluation of Dose Concordance for Transcriptional and Apical Responses
For each gene expression dataset, BMD values for each GO biological process class were calculated for each tissue and each of the 48 combinations of pre-modeling probe filtering, BMD modeling, and gene set summarization parameters. The lowest median BMD value among GO biological processes in either tissue (liver and kidney) was used as the transcriptomic BMD. The log10-transformed transcriptomic BMD values were then compared with the minima of the log10-transformed chronic non-cancer and cancer apical BMD values using Pearson’s correlation coefficient and RMSD. For chemicals with replicate transcriptomic studies, the transcriptomic BMD values within each study were averaged together to derive a single transcriptomic BMD estimate for comparison to the apical BMD. The RMSD was calculated as follows:
Where Xi is the log10 transcriptomic BMD value for the ith chemical, Yi is the minimum of the log10 chronic non-cancer and cancer apical BMD values for the ith chemical, and N is the total number of chemicals. Combinations that failed to derive a transcriptomic BMD (i.e., that had no GO biological process class passing all filters in either tissue) for any of the 14 chemicals were removed. The combinations of parameters were rank ordered based on RMSD.
Across all combinations of parameters that successfully derived a transcriptomic BMD for all 14 chemicals, the Pearson correlation coefficient ranged from 0.804 to 0.917, while the RMSD ranged from 0.567 to 0.958 (log10 mg/kg-day). The top five combinations of parameters based on the RMSD are provided in Table 4-3. In each of the top five combinations, an absolute fold-change >1.5, BMD/BMDL < 20, and minimum of 3 genes per GO class were consistently represented. Only the William’s p-value and percentage of genes in the set varied in their representation in the top five ranked combinations.
Table 4-3
Top five combinations of pre-modeling probe filter, BMD modeling, and gene set summarization parameters based on RMSD.
The best overall combination of parameters based on the minimum RMSD of the transcriptomic versus apical BMD values includes pre-modeling probe filtering criteria of |Fold-Change| > 1.5 with a William’s trend test p-value of <0.05 and a post-modeling filter to remove probes with BMD/BMDL ratio > 20. When summarizing the results for the rat S1500+ assay based on GO biological process class, the best combination of parameters had a minimum of 3 genes with a valid BMD as the cutoff, with no minimum requirement on percent coverage. Using the recommended parameter combinations, the Pearson correlation coefficient and RMSD of the transcriptomic versus chronic apical BMD values were 0.910 and 0.567, respectively (Figure 4-2). The median absolute ratio of the transcriptomic BMD and chronic non-cancer apical BMD values was 3.2 ± 1.9 (MAD). The maximum absolute fold-difference was 7.87. The RMSD value for the best parameter combination was slightly higher than that reported by Johnson and colleagues when comparing transcriptomic and apical POD values for 29-day toxicity studies [0.54 and 0.48 for consistent and inconsistent dose levels, respectively; (Johnson et al. 2020)]. For further comparison, the RMSD value is similar to the range of inter-study standard deviation estimates for the LOAELs for systemic toxicity in repeated dose studies, approximated as residual RMSE in log10-mg/kg-day units [0.45-0.56; (Pham et al. 2020)]. The results suggest that the error associated with the concordance between the transcriptomic BMD values versus non-cancer and cancer apical BMD values for the best parameter combination is approximately equivalent to the inter-study variability in the repeated dose toxicity study itself.
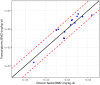
Figure 4-2
Scatter plot of log10 transcriptomic BMD versus the minimum of the chronic non-cancer and cancer apical log10 BMD values for the top ranked combination of pre-modeling probe filter, BMD modeling, and gene set summarization parameters (Table 4-3). The (more...)
Concordance between the transcriptomic and chronic apical BMDL values were also compared. Using the best parameter combination, the Pearson correlation coefficient and RMSD of the transcriptomic versus chronic apical BMDL values were 0.908 and 0.694, respectively (Figure 4-3). The median absolute fold-difference between the transcriptomic BMDL and chronic apical BMDL values was 2.8 ± 1.6 (MAD). Notably, the majority of transcriptomic BMDL values are lower than the chronic apical BMDL values. This contrasts with the BMD values where the transcriptomic BMD values were approximately equally distributed above and below the unity line. This suggests that the confidence intervals for the transcriptomic BMD values are slightly wider than those for the apical BMD values leading to more conservative PODs used to derive reference values.
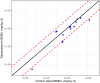
Figure 4-3
Scatter plot of log10 transcriptomic BMDL versus the minimum of the chronic non-cancer and cancer apical log10 BMDL values for the top ranked combination of pre-modeling probe filter, BMD modeling, and gene set summarization parameters (Table 4-3). The (more...)
4.2.2. EVALUATION OF INTER-STUDY REPRODUCIBILITY
4.2.2.1. Overview
To evaluate the inter-study reproducibility of the transcriptomic BMD and BMDL values, the transcriptomic data from three independently replicated chemicals were analyzed. The replicated chemicals included furan (n = 3), perfluorooctanoic acid (n = 3), and bromodichloroacetic acid (n = 3). The replicate studies were run at the same contract lab over the course of several years and required the preparation of new dosing solutions for each study using the same supplier and lot for each chemical.
4.2.2.2. Calculation of Inter-Study Reproducibility
The raw sequencing reads (FASTQ files) for the three independently replicated chemicals with chronic rodent bioassays were obtained from the NTP. The detailed methods for aligning, normalizing, and quality control of the sequencing data are outlined in the Appendix (Section 6.2). Following quality control, the dose response series for each of the replicates were analyzed using the complete transcriptomic dose response analysis process including: 1) pre-modeling dataset evaluation to determine adequate signal; 2) pre-modeling probe filtering to remove those that are not responding to treatment; 3) dose response modeling of the individual probes, identifying the best-fit model, and deriving BMD(L) values; and 4) combining the individual probes into gene sets and summarizing the transcriptional BMD(L) values. For the pre-modeling dataset evaluation, an ANOVA evaluation with an FDR < 0.05 cut-off for 1 or more probes was used. For the rest of the dose response modeling process, the top 5 combinations of pre-modeling probe filtering, BMD modeling, and gene set summarization parameters in Table 4-3 were evaluated. The inter-study reproducibility was calculated using the estimated standard deviation (SD) of the transcriptomic BMD and BMDL values. The SD was estimated as follows:
Where Xi and Yi are the log10 transcriptomic BMD or BMDL values for the ith pair of independently replicated studies for the same chemicals, and N is the total number of pairs of independently replicated studies.
The results suggest that the parameter combinations which produced transcriptomic BMDs that were most concordant with the apical BMDs also produce transcriptional BMDs that are highly reproducible based on the independent replicate studies for the three chemicals (Table 4-4). Furthermore, all five of the parameter combinations result in highly similar SD values for both the transcriptional BMD and BMDL values. Therefore, we chose the parameter combination with the overall lowest RMSD for transcriptional versus apical BMD values, as the differences in BMD/BMDL between the top combinations were negligible.
Table 4-4
Inter-study variability of the median BMD and BMDL values for the top five combinations of pre-modeling probe filter, BMD modeling, and gene set summarization parameters.
4.2.3. EVALUATION OF FAMILY-WISE ERROR RATE
4.2.3.1. Overview
To estimate the family-wise error rate, individual samples from the corn oil vehicle control groups were randomly distributed into 1,000 sham dose response series for each tissue. The sham dose response series were then analyzed to estimate the family-wise error rate for the pre-modeling dataset evaluation step as well as the complete transcriptomic dose response analysis process.
4.2.3.2. Pre-Modeling Dataset Evaluation of Sham Dose Response Series
According to EPA benchmark dose guidance, the dataset being modeled should have a statistically or biologically significant dose-related trend (EPA 2012). For transcriptomic dose response data, the NTP report recommended performing an ANOVA prior to dose response modeling with a cut-off of at least one gene or probe showing statistical significance at a Benjamini-Hochberg FDR corrected p-value < 0.05 (NTP 2018). To evaluate the fitness of this recommendation for the specific study design and transcriptomics platform employed in the ETAP, the raw sequencing reads (FASTQ files) for the corn oil vehicle control groups were obtained from NTP. The methods for aligning, normalizing, and quality control of the sequencing data are outlined in the Appendix (Section 6.2). A subset of 53 liver and kidney samples from the corn oil vehicle control groups (14 studies that used corn oil as the matched vehicle control X 4 animals per study minus low quality and outlier samples) were randomly distributed into 1,000 sham dose response series for each tissue. Each sham dose response series consisted of nine groups (one control group and eight mock positive dose groups) with four samples per group. To represent realistic dose ranges, an equal fraction of sham dose response series was assigned the 8 lowest doses for each of the 14 chemical regimens tested. The sham dose response series were analyzed using ANOVA and a range of FDR corrected p-values. The estimated family-wise error rate was computed based on the number of the 1,000 sham dose response series with at least one probe passing the ANOVA with FDR correction (Fig. 4-4). The results suggest that a pre-modeling ANOVA evaluation with an FDR < 0.05 cut-off for 1 or more probes results in an estimated family-wise error rate of 0.046 in both the liver and kidney. The estimated family-wise error rate for the sham dose response series approximates the target FDR for the analysis.
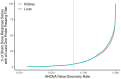
Figure 4-4
Percentage of sham dose response datasets with at least one probe showing statistical significance based on an ANOVA with a p-value of varying FDR correction. The sham dose response datasets were created by randomly distributing the individual corn oil (more...)
4.2.3.3. Complete Transcriptomic Dose Response Analysis of the Sham Dose Response Series
Although characterizing the family-wise error rate of the initial pre-modeling dataset evaluation step provides an understanding of the number of potential datasets that may make it into the dose response modeling process erroneously, the results may not reflect the overall family-wise error rate of the complete transcriptomic dose response analysis process. To estimate the overall family-wise error rate for identifying a gene set-level BMD with the specific study design and transcriptomics platform employed in the ETAP, the same set of 1,000 sham dose response series were analyzed using the complete transcriptomic dose response analysis process including: 1) pre-modeling dataset evaluation to determine adequate signal; 2) pre-modeling probe filtering to remove those that are not responding to treatment; 3) dose response modeling of the individual probes, identifying the best-fit model, and deriving a BMD value together with its lower confidence bound (i.e., BMD and BMDL); and 4) combining the individual probes into gene sets and summarizing the transcriptional BMD and BMDL values. For the pre-modeling dataset evaluation, an ANOVA evaluation with an FDR < 0.05 cut-off for 1 or more probes was used. For the rest of the dose response modeling process, the top 5 combinations of pre-modeling probe filter, BMD modeling, and gene set summarization parameters in Table 4-3 were evaluated. A false positive was counted when a sham dose response series had at least one GO biological process class with a valid BMD and BMDL. The results suggest that combining the pre-modeling dataset evaluation step with the transcriptomic dose response modeling process significantly reduced the family-wise error rate from 0.046 to less than 0.01. All of the top five combinations of pre-modeling probe filter, BMD modeling, and gene set summarization parameters have an overall family-wise error rate of less than 0.01 with the highest ranked combination of parameters with a family-wise error rate of 0.006 (Table 4-5).
Table 4-5
Overall family-wise error rate of the top five combinations of pre-modeling probe filter, BMD modeling, and gene set summarization parameters.
4.3. COMPARISON OF TRANSCRIPTIONAL AND APICAL DOSE CONCORDANCE IN THE CONTEXT OF INTER-STUDY VARIABILITY
The evaluation of the concordance between transcriptional BMD values from the short-term in vivo studies and the apical BMD values from the chronic rodent bioassays are confounded by the inter-study variability in both dimensions. Estimating and explicitly considering this variability is important for interpreting the concordance metrics (e.g., RMSD) and the level of confidence in the application of the ETAP. To provide this context, the concordance MSD of the top combination of pre-modeling probe filter, BMD modeling, and gene set summarization parameters was compared with an estimate of the lower bound of the expected MSD given inter-study variances.
4.3.1. DERIVATION OF MSD LOWER BOUND
Let Xc be the observed transcriptomic BMD (log10 mg/kg-day) and Yc the observed apical BMD (log10 mg/kg-day) for chemical c, where study design was standardized across chemicals. Following the work of Pham and colleagues (Pham et al. 2020), the apical BMD values were assumed to be random variables with means dependent on chemical and study design (but note study design is standardized across chemicals in this study) and constant variance after accounting for chemical and study design (i.e., common variance across chemicals). In the absence of evidence to the contrary, the same was assumed for the transcriptomic BMD values. That is, it was assumed that E[Xc] = μX(c) and E[Yc] = μY(c), where μX(c) and μY(c) are the mean transcriptomic and apical BMD values for chemical c, respectively; and and are the inter-study, within-chemical variances for transcriptomic and apical BMD values, respectively.
Let Zc = Xc – Yc be the difference between observed transcriptomic and apical BMD values for chemical c. Then E[Zx] = μZ = μX(c) − μY(c) (note that the difference in BMD means was assumed to be constant across chemicals) and (note that Xc and Yc are conditionally independent given chemical means, so no covariance term is needed).
The MSD concordance statistic between Xc and Yc for n chemicals is an unbiased estimator of :
4.3.2. ESTIMATES OF INTER-STUDY VARIANCES AND LOWER BOUND OF EXPECTED CONCORDANCE MSD
Inter-study replicates were used to estimate the transcriptomic BMD variance, . For replicates i and j of chemical c, E[Xc,i − Xc,j] = 0 and from Section 4.3.1 above. Let k be the number of chemicals with replicate transcriptomic BMD estimates, let rc be the number of observed replicates for chemical c, and let Ic = {1,2,…,rc}. An unbiased estimator of is:
4.3.3. MSD OF THE TOP COMBINATION OF TRANSCRIPTOMIC DOSE RESPONSE MODELING PARAMETERS COMPARED WITH LOWER BOUND OF THE EXPECTED CONCORDANCE MSD GIVEN INTER-STUDY VARIANCES
The MSD of the top combination of pre-modeling probe filter, BMD modeling, and gene set summarization parameters using mean log10 BMD values for chemicals that had replicates was 0.5672=0.321 (log10 mg/kg-day)2. However, using mean BMD values for only some chemicals violates the assumption of equal variance across chemicals used to derive the lower bound of expected MSD. For comparison with the lower bound estimate, the concordance MSD for the top model was computed using all combinations of single replicates per chemical, and the minimum and maximum of these point estimates were 0.285 and 0.386, respectively. Thus, the full range of MSD values computed using the single chemical replicates falls within the range of lower bound estimates for expected concordance MSD of 0.267 - 0.617 when considering the inter-study variances for both the transcriptomic studies and repeated dose studies examining systemic effects. The results suggest that the error associated with the concordance between the transcriptomic BMD values versus non-cancer and cancer apical BMD values is approximately equivalent to the combined inter-study variability associated with the 5-day transcriptomic study and the chronic rodent bioassay.
Footnotes
- 26
The second NTP dataset is available at: https://doi
.org/10.22427 /NTP-DATA-002-00099-0001-000-1 - 27
NTP histopathological data for tris(2-chloropropyl) phosphate: https://ntp
.niehs.nih.gov/go/TS-m20263
- DEVELOPMENT OF TRANSCRIPTOMIC POINTS OF DEPARTURE FOR ETAP - Scientific Studies ...DEVELOPMENT OF TRANSCRIPTOMIC POINTS OF DEPARTURE FOR ETAP - Scientific Studies Supporting Development of Transcriptomic Points of Departure for EPA Transcriptomic Assessment Products (ETAPs)
Your browsing activity is empty.
Activity recording is turned off.
See more...