Summary
Clinical characteristics.
Aicardi syndrome is a neurodevelopmental disorder that affects primarily females. Initially it was characterized by a typical triad of agenesis of the corpus callosum, central chorioretinal lacunae, and infantile spasms. As more affected individuals have been ascertained, it has become clear that not all affected girls have all three features of the classic triad and that other neurologic and systemic defects are common, including other brain malformations, optic nerve abnormalities, other seizure types, intellectual disability of varying severity, and scoliosis.
Diagnosis/testing.
The diagnosis of Aicardi syndrome is based exclusively on clinical findings. Modified diagnostic criteria include either presence of the classic triad or the presence of two of the classic triad plus at least two other major or supporting features. A gene for Aicardi syndrome has not been identified, but several observations support a hypothesis that Aicardi syndrome is caused by de novo pathogenic variants in a gene on the X chromosome that is subject to X-chromosome inactivation.
Management.
Treatment of manifestations: Treatment is individualized and long-term management by a pediatric neurologist with expertise in management of infantile spasms and medically refractory epilepsy is essential. Physical therapy, occupational therapy, and speech therapy should begin at the time of diagnosis. Appropriate musculoskeletal support and treatment for prevention of scoliosis-related complications is indicated.
Surveillance: Includes routine monitoring of growth, nutritional status and safety of oral intake, seizure control, developmental progress and educational needs, respiratory function and aspiration risk, and the spine and degree of scoliosis.
Genetic counseling.
With the exception of affected monozygotic twin girls, all individuals with Aicardi syndrome reported to date have represented simplex cases (i.e., a single affected family member). Parent-to-child transmission of Aicardi syndrome has not been reported, and the recurrence risk to sibs is thought to be less than 1%. While prenatal ultrasound or intrauterine MRI findings of corpus callosum agenesis and neuronal migration abnormalities may be suggestive of Aicardi syndrome, no findings are diagnostic.
Diagnosis
No consensus clinical diagnostic criteria for Aicardi syndrome have been published.
The diagnosis of Aicardi syndrome is based exclusively on clinical findings. Modified diagnostic criteria have been proposed [Sutton et al 2005, adapted from Aicardi 1999]:
- The presence of the classic triad is diagnostic for Aicardi syndrome.
- The presence of two of the classic triad plus at least two other major or supporting features is strongly suggestive of the diagnosis of Aicardi syndrome.
Classic triad
- Agenesis of the corpus callosum
- Distinctive chorioretinal lacunae
- Infantile spasms
Major features
- Cortical malformations (mostly polymicrogyria)
- Periventricular and subcortical heterotopia
- Cysts around third cerebral ventricle and/or choroid plexus
- Optic disc/nerve coloboma or hypoplasia
Supporting features
- Vertebral and rib abnormalities
- Microphthalmia
- "Split-brain" EEG
- Gross cerebral hemispheric asymmetry
- Vascular malformations or vascular malignancy
Note: Aicardi syndrome appears to be an X-linked dominant disorder with lethality in males; however, no gene or candidate region on the X chromosome has been identified (see Molecular Pathogenesis).
Clinical Characteristics
Clinical Description
Aicardi syndrome, first described by Aicardi et al [1965], is a neurodevelopmental disorder that affects primarily females [Aicardi 1999, Van den Veyver 2002, Aicardi 2005]. Initially it was characterized by a typical triad of agenesis of the corpus callosum, typical chorioretinal lacunae, and infantile spasms; however, as more affected individuals have been ascertained, it has become clear that other neurologic and systemic defects are common. Indeed, not all affected girls have all three features of the classic triad (see Table 1).
Table 1.
Select Features of Aicardi Syndrome
Neurologic. The neurologic examination can reveal microcephaly, axial hypotonia, and appendicular hypertonia with spasticity often affecting one side and brisk deep tendon reflexes as well as hemiparesis [Aicardi 2005; Author, unpublished data]. Moderate-to-severe developmental delay and intellectual disability are typical, although individuals with only mild or no learning disabilities or developmental delay have been reported (reviewed in Wong & Sutton [2018]).
Many girls with Aicardi syndrome develop seizures before age three months, and most before age one year. Infantile spasms are seen early on; ongoing medically refractory epilepsy with a variety of seizure types develops over time. Common EEG findings include asynchronous multifocal epileptiform abnormalities with burst suppression and dissociation between the two hemispheres.
Brain MRI reveals dysgenesis of the corpus callosum, which is most often complete but can be partial. Polymicrogyria or pachygyria, which are predominantly frontal and perisylvian and associated with underopercularization, are typical. Periventricular and intracortical gray matter heterotopia are very common. Gross cerebral asymmetry, choroid plexus papillomas, ventriculomegaly, and intracerebral cysts, often at the third ventricle and in the choroid plexus, are frequently present [Aicardi 2005, Hopkins et al 2008]. Recently, posterior fossa and cerebellar abnormalities are increasingly recognized as important components of the phenotype [Hopkins et al 2008, Steffensen et al 2009].
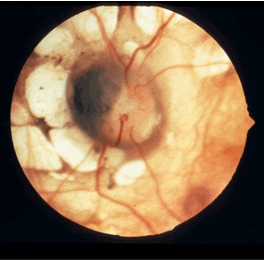
Ophthalmologic. The pathognomonic chorioretinal lacunae of Aicardi syndrome are white or yellow-white, well-circumscribed, round, depigmented areas of the retinal pigment epithelium and underlying choroid with variably dense pigmentation at their borders (Figure 1) [Fruhman et al 2012] that can cluster in the posterior pole of the globe around the optic nerve. The sensory retina overlying the lacunae is usually intact but may be disorganized or entirely absent.
Figure 1.
Classic lacunae surround the modestly dysplastic left optic disc. Note the nasal "papilla nigra" appearance and the anomalous branching patterns of the central vasculature. Image courtesy of Dr Richard A Lewis
Almost all individuals have some ocular abnormalities. Fruhman et al [2012] described data on 40 females with Aicardi syndrome examined by a single expert ophthalmologist and retinal specialist. Of the 75 eyes that could be evaluated, all had chorioretinal lacunae. Other common optic nerve findings included:
- Coloboma (39% of eyes)
- Glial proliferation (19% of eyes)
- Severe dysplasia (17% of eyes)
- Pseudoadenomatous proliferation of the retinal pigment epithelium (14% of eyes)
All eye findings can be unilateral or bilateral and asymmetric.
Craniofacial. Characteristic facial features reported in Aicardi syndrome include a short philtrum, prominent premaxilla with resultant upturned nasal tip and decreased angle of the nasal bridge, large ears, and sparse lateral eyebrows [Sutton et al 2005].
Plagiocephaly and facial asymmetry, occasionally with cleft lip and palate (3%), have been reported. Pierre Robin sequence has been reported in a single case [Jensen & Christiansen 2004].
Skeletal. Costovertebral defects, such as hemivertebrae, block vertebrae, fused vertebrae, and missing ribs, are common and can lead to marked scoliosis in up to one third of affected individuals [Donnenfeld et al 1989]. Hip dysplasia has been reported.
Gastrointestinal. Constipation, gastroesophageal reflux, diarrhea, and feeding difficulties are perceived by parents to be the second most difficult problem to manage (after seizures) [Glasmacher et al 2007].
Extremities. Small hands, along with an increased incidence of hand malformations, have been reported [Sutton et al 2005].
Dermatologic. An increased incidence of vascular malformations and pigmentary lesions has been observed [Sutton et al 2005].
Tumors/malignancies. The incidence of tumors may be increased. A variety of rare tumor types have been reported: benign tumors such as choroid plexus papillomas [Taggard & Menezes 2000, Pianetti Filho et al 2002] and lipomas, as well as malignant tumors including angiosarcomas, hepatoblastomas, meduloblastoma, embryonal carcinomas, and teratomas [Sutton et al 2005, Wharton et al 2018].
Growth. The average heights and weights of girls with Aicardi syndrome closely follow those of the general population up to ages seven and nine years, respectively, after which the growth rate for both height and weight is lower. Growth curves for Aicardi syndrome based on parent survey data have been published [Glasmacher et al 2007]. The weight-versus-height ratio remains similar to the general population.
Endocrine. Either precocious puberty or delayed puberty may be present [Glasmacher et al 2007].
Survival. Survival in Aicardi syndrome is highly variable and likely depends on the severity of seizures. In a survey by Glasmacher et al [2007], the mean age at death was 8.3 years, although the median age of death was 18.5 years. The ages of death were distributed from before age one year to older than 23 years. The oldest surviving individual reported in this survey was age 32 years. Another paper also indicated that the survival is longer than previously reported, especially in more mildly affected individuals, with the highest risk of death at age 16 years and a probability of survival at age 27 years of 0.62 [Kroner et al 2008].
Nomenclature
Note: Aicardi syndrome is NOT related to Aicardi-Goutières syndrome, an early-onset encephalopathy.
Prevalence
Aicardi syndrome is very rare and appears to affect all ethnicities equally. Its incidence has been estimated at between 1:105,000 and 1:167,000 in the United States and between 1:93,000 and 1:99,000 in some European countries [Kroner et al 2008].
The exact prevalence of Aicardi syndrome is unknown; it has been estimated to be at least 853 in the US and over 4,000 worldwide [Kroner et al 2008].
Differential Diagnosis
Agenesis of the corpus callosum may occur in isolation, in conjunction with other brain malformations, or as part of a larger syndrome. It has been suggested that agenesis of the corpus callosum in association with cysts that do not communicate with the ventricles and presence of subependymal heterotopia and polymicrogyria is relatively specific for Aicardi syndrome [Barkovich et al 2001].
Neuronal migration disorders, including polymicrogyria, pachygyria, and heterotopia, may occur as isolated malformations or as part of the phenotype associated with other syndromes or chromosome abnormalities.
Infantile spasms are observed in most girls with Aicardi syndrome, but this type of seizure is not specific for Aicardi syndrome. Infantile spasms may occur in isolation or as part of the phenotype of other syndromes, inborn errors of metabolism, or chromosome disorders.
Hereditary disorders. See Table 2.
Table 2.
Genes of Interest in the Differential Diagnosis of Aicardi Syndrome
Syndromes of unknown cause
- Orofaciodigital syndrome type IX (OFD9; OMIM 258865). Although chorioretinal lacunae are virtually pathognomonic for Aicardi syndrome, they have also been reported in OFD .
- Oculocerebrocutaneous syndrome (OMIM 164180) characterized by orbital cysts and anophthalmia or microphthalmia, focal skin defects, brain malformations that include polymicrogyria, periventricular nodular heterotopias, enlarged lateral ventricles, and agenesis of the corpus callosum, is predominant in males and has a pathognomonic mid-hindbrain malformation [Moog et al 2005].
Management
No clinical practice guidelines for Aicardi syndrome have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Aicardi syndrome, the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with Aicardi Syndrome
Treatment of Manifestations
Table 4.
Treatment of Manifestations in Individuals with Aicardi Syndrome
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Communication Issues
Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Social/Behavioral Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder (ADHD), when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Surveillance
Table 5.
Recommended Surveillance for Individuals with Aicardi Syndrome
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to genetic counseling of relatives at risk.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
The gene(s) in which pathogenic variants are causative of Aicardi syndrome are not known. While autosomal inheritance cannot be fully excluded in the absence of a known genetic cause, several observations support a hypothesis that Aicardi syndrome is caused by a de novo pathogenic variant in a gene on the X chromosome that is subject to X-chromosome inactivation (see Molecular Genetics).
Aicardi syndrome is typically seen in females but has also been reported in males with a 47,XXY karyotype and, very rarely, in 46,XY males.
Risk to Family Members
Parents of a proband. With the exception of affected monozygotic twin girls, all individuals with Aicardi syndrome reported to date have represented simplex cases (i.e., a single affected family member). Parent-to-child transmission of Aicardi syndrome has not been reported.
Sibs of a proband. The recurrence risk to sibs is thought to be less than 1%.
Offspring of a proband
- No instances of mother-to-daughter transmission have been documented, despite the presence of rare adult women with Aicardi syndrome who do not have a degree of intellectual disability that would result in decreased reproductive fitness. This raises the question of whether fertility may be reduced in women with Aicardi syndrome. The hypothesis that fertility in Aicardi syndrome is reduced could also be supported by reports of precocious puberty in individuals with Aicardi syndrome [Glasmacher et al 2007], which reflect an underlying reproductive hormone dysregulation that could lead to both precocious puberty and inability to sustain a pregnancy successfully.
- While the genetic cause is currently unknown, theoretically, if a female with Aicardi syndrome conceives, the risk that the causative variant will be transmitted could be as high as 50%.
Other family members. Given that almost all probands with Aicardi syndrome reported to date have represented simplex cases, the risk to other family members is presumed to be low.
Related Genetic Counseling Issues
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing
Molecular genetic testing. Prenatal testing by molecular genetic testing is not possible as the gene(s) in which pathogenic variants are causative of Aicardi syndrome are not known.
Other
- Some features detected on prenatal ultrasound examination, such as agenesis of the corpus callosum with intracranial cysts in a female fetus, may raise suspicion for Aicardi syndrome and a number of other developmental brain abnormalities.
- Findings of corpus callosum agenesis and neuronal migration abnormalities on intrauterine MRI may be suggestive but not diagnostic [Masnada et al 2020].
- Other features, such as costovertebral defects and microphthalmia, are more difficult to detect prenatally and are not present in all cases.
- Definitive prenatal diagnosis of Aicardi syndrome has not been reported in a low-risk pregnancy; furthermore, definitive diagnosis of Aicardi syndrome relies on neonatal confirmation of suspected findings and detection of additional features including chorioretinal lacunae and seizures.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Aicardi Syndrome FoundationPO Box 3202St. Charles IL 60174Phone: 800-374-8518 (toll-free)Email: web@aicardisyndrome.org
- MedlinePlus
- National Institute of Neurological Disorders and Stroke (NINDS)PO Box 5801Bethesda MD 20824Phone: 800-352-9424 (toll-free); 301-496-5751; 301-468-5981 (TTY)
- Aicardi Syndrome International RegistryPhone: 301-230-4674Email: byk@rti.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table B.
OMIM Entries for Aicardi Syndrome (View All in OMIM)
Molecular Pathogenesis
A gene for Aicardi syndrome has not been identified, but several observations support a hypothesis that Aicardi syndrome is caused by de novo pathogenic variants in a gene on the X chromosome that is subject to X-chromosome inactivation [Van den Veyver 2002].
- Nearly all affected individuals are female and, except for one pair of sisters and a pair of concordant monozygotic twins, all reported cases are simplex (i.e., a single occurrence in a family).
- At least six pairs of twins who are discordant for Aicardi syndrome are known, five of whom are confirmed dizygotic, excluding the possibility that the etiology is a prenatal toxin or other disruptive event [Taggard & Menezes 2000].
- Rare known males with a confirmed diagnosis of Aicardi syndrome have a 47,XXY karyotype [Aicardi 1999, Chen et al 2009, Zubairi et al 2009, Shetty et al 2014]. The presence of Aicardi syndrome in males with a 46,XY karyotype has been disputed, but new cases have been reported. It is possible that these are caused by mosaic pathogenic variants in the putative Aicardi syndrome gene [Chappelow et al 2008, Anderson et al 2009]
- The variable severity and asymmetry of the Aicardi syndrome phenotype could be explained if the putative mutated gene undergoes X-chromosome inactivation. Earlier limited studies, some using older methods, had conflicting results, showing either random or skewed X-chromosome inactivation patterns. The most recent study on 35 individuals with Aicardi syndrome, the largest series examined thus far, demonstrated non-random X-chromosome inactivation in leukocyte-derived DNA in 11 of 33 informative individuals, with six (18%) having extremely skewed patterns. This is significantly increased compared to the general population [Eble et al 2009].
- Because a subset of the clinical findings of Aicardi syndrome (including colobomas, agenesis of the corpus callosum, microphthalmia, and seizures) overlaps with those of microphthalmia with linear skin defects (MLS) syndrome and with Goltz syndrome (focal dermal hypoplasia), it was hypothesized that these three conditions were allelic and that the gene for Aicardi syndrome was located on Xp22. However, sequencing and deletion studies now indicate that Aicardi syndrome is likely not allelic to MLS syndrome [Van den Veyver 2002] or to Goltz syndrome [Wang et al 2007].
- Until the genetic basis of Aicardi syndrome is known, the possibility remains that Aicardi syndrome is caused by a de novo pathogenic variant on an autosome with sex-limited expression in females. Interestingly, while one report described a girl with an 11-Mb deletion of 1p36 with an Aicardi syndrome phenocopy [Bursztejn et al 2009], it is important to note that she did not have the chorioretinal lacunae typical of Aicardi syndrome and did have facial features and cardiac defects characteristic of the 1p36 deletion syndrome (OMIM 607872).
Efforts to identify the mutated gene using array comparative hybridization with genome-wide DNA microarrays [Wang et al 2009] and X-chromosome DNA microarrays [Yilmaz et al 2007] have not been successful to date. Exome and X-chromosome genome sequencing by at least three separate groups have also been unsuccessful [Author, unpublished data].
Chapter Notes
Acknowledgments
Research by the authors is supported by the Aicardi Syndrome Foundation and the Baylor College of Medicine Intellectual and Developmental Disabilities Research Center, supported by IDDRC grant number U54HD083092 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health & Human Development or the National Institutes of Health.
The retina photo and legend were provided by Dr Richard A Lewis.
Revision History
- 12 November 2020 (bp) Comprehensive update posted live
- 6 November 2014 (me) Comprehensive update posted live
- 27 April 2010 (me) Comprehensive update posted live
- 30 June 2006 (ca) Review posted live
- 2 August 2005 (ivv) Original submission
References
Literature Cited
- Aicardi J. Aicardi Syndrome: Old and new findings. Internat Pediatr. 1999;14:5–8.
- Aicardi J. Aicardi syndrome. Brain Dev. 2005;27:164–71. [PubMed: 15737696]
- Aicardi J, Levebre J, Lerique-Koechlin A. A new syndrome: Spasms in flexion, callosal agenesis, ocular abnormalities. Electroencephalogr Clin Neurophysiol. 1965;19:609–10.
- Anderson S, Menten B, Kogelenberg M, Robertson S, Waginger M, Mentzel HJ, Brandl U, Skirl G, Willems P. Aicardi syndrome in a male patient. Neuropediatrics. 2009;40:39–42. [PubMed: 19639527]
- Bursztejn AC, Bronner M, Peudenier S, Gregoire MJ, Jonveaux P, Nemos C. Molecular characterization of a monosomy 1p36 presenting as an Aicardi syndrome phenocopy. Am J Med Genet A. 2009;149A:2493–500. [PubMed: 19842196]
- Chappelow AV, Reid J, Parikh S, Traboulsi EI. Aicardi syndrome in a genotypic male. Ophthalmic Genet. 2008;29:181–3. [PubMed: 19005990]
- Barkovich AJ, Simon EM, Walsh CA. Callosal agenesis with cyst: a better understanding and new classification. Neurology. 2001;56:220–7. [PubMed: 11160959]
- Chen TH, Chao MC, Lin LC, Jong YJ, Yang SN, Lai YH, Chen HL. Aicardi syndrome in a 47, XXY male neonate with lissencephaly and holoprosencephaly. J Neurol Sci. 2009;278:138–40. [PubMed: 19155022]
- Devinsky O, Verducci C, Thiele EA, Laux LC, Patel AD, Filloux F, Szaflarski JP, Wilfong A, Clark GD, Park YD, Seltzer LE, Bebin EM, Flamini R, Wechsler RT, Friedman D. Open-label use of highly purified CDB (Epidolex®) in patients with CDKL5 deficiency disorder and Aicardi, Dup 15q, and Doose syndromes. Epilepsy Behav. 2018;86:131–7. [PubMed: 30006259]
- Donnenfeld AE, Packer RJ, Zackai EH, Chee CM, Sellinger B, Emanuel BS. Clinical, cytogenetic, and pedigree findings in 18 cases of Aicardi syndrome. Am J Med Genet. 1989;32:461–7. [PubMed: 2773986]
- Eble TN, Sutton VR, Sangi-Haghpeykar H, Wang X, Lewis RA, Van den Veyver IB. X-chromosome inactivation in Aicardi Syndrome. Hum Genet. 2009;125:211–6. [PMC free article: PMC2660246] [PubMed: 19116729]
- Fruhman G, Eble TN, Gambhir N, Sutton VR, Van den Veyver IB, Lewis RA. Ophthalmologic findings in Aicardi syndrome. J AAPOS. 2012;16:238–41. [PMC free article: PMC3650611] [PubMed: 22681940]
- Glasmacher MA, Sutton VR, Hopkins B, Eble T, Lewis RA, Park Parsons D, Van den Veyver IB. Phenotype and management of Aicardi syndrome: new findings from a survey of 69 children. J Child Neurol. 2007;22:176–84. [PubMed: 17621479]
- Hopkins B, Sutton VR, Lewis RA, Van den Veyver I, Clark GD. Neuroimaging aspects of Aicardi syndrome. Am J Med Genet A. 2008;146A:2871–8. [PMC free article: PMC2597151] [PubMed: 18925666]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Jensen AA, Christiansen SP. Aicardi syndrome with Pierre Robin sequence. J AAPOS. 2004;8:187–9. [PubMed: 15088056]
- Kroner BL, Preiss LR, Ardini MA, Gaillard WD. New incidence, prevalence, and survival of Aicardi syndrome from 408 cases. J Child Neurol. 2008;23:531–5. [PubMed: 18182643]
- Masnada S, Chiara D, Giana I, Manuela F, Marco S, Andrea A, Patrizia A, Nadia BB, Valeria C, Mara C, Bernardo DB, Francesca D, Valentina G, Elisa FRL, Carlo F, Lucio G, Simona O, Lorenzo P, Erika R, Angonio R, Mariasavina S, Carlotta S, Pierangelo V, Anna P, Andrea R, Cecila P. Aicardi syndrome: Key fetal MRI features and prenatal differential diagnosis. Neuropediatrics. 2020;51:276–85. [PubMed: 32620025]
- Moog U, Jones MC, Bird LM, Dobyns WB. Oculocerebrocutaneous syndrome: the brain malformation defines a core phenotype. J Med Genet. 2005;42:913–21. [PMC free article: PMC1735958] [PubMed: 15879499]
- Pianetti Filho G, Fonseca LF, da Silva MC. Choroid plexus papilloma and Aicardi syndrome: case report. Arq Neuropsiquiatr. 2002;60:1008–10. [PubMed: 12563397]
- Shetty J, Fraser J, Goudie D, Kirkpatrick M. Aicardi syndrome in a 47 XXY male - a variable developmental phenotype? Eur J Paediatr Neurol. 2014;18:529–31. [PubMed: 24657013]
- Steffensen TS, Gilbert-Barness E, Lacson A, Margo CE. Cerebellar migration defects in aicardi syndrome: an extension of the neuropathological spectrum. Fetal Pediatr Pathol. 2009;28:24–38. [PubMed: 19116813]
- Sutton VR, Hopkins BJ, Eble TN, Gambhir N, Lewis RA, Van den Veyver IB. Facial and physical features of Aicardi syndrome: infants to teenagers. Am J Med Genet A. 2005;138A:254–8. [PubMed: 16158440]
- Taggard DA, Menezes AH. Three choroid plexus papillomas in a patient with Aicardi syndrome. A case report. Pediatr Neurosurg. 2000;33:219–23. [PubMed: 11124640]
- Van den Veyver IB. Microphthalmia with linear skin defects (MLS), Aicardi, and Goltz syndromes: are they related X-linked dominant male-lethal disorders? Cytogenet Genome Res. 2002;99:289–96. [PubMed: 12900577]
- Wang X, Reid Sutton V, Omar Peraza-Llanes J, Yu Z, Rosetta R, Kou YC, Eble TN, Patel A, Thaller C, Fang P, et al. Mutations in X-linked PORCN, a putative regulator of Wnt signaling, cause focal dermal hypoplasia. Nat Genet. 2007;39:836–8. [PubMed: 17546030]
- Wang X, Sutton VR, Eble TN, Lewis RA, Gunaratne P, Patel A, Van den Veyver IB. A genome-wide screen for copy number alterations in Aicardi syndrome. Am J Med Genet A. 2009;149A:2113–21. [PMC free article: PMC3640635] [PubMed: 19760649]
- Wharton JD, Johnson S, Connelly JA, Hills T, Gingles L, Wood M, Crane GL, Katzenstein HM. High-dose chemotherapy is efficacious and well tolerated in a toddler with Aicardi syndrome and malignant Sacrococcygeal teratoma. J Pediatr Hematol Oncol. 2018;40:e467–9. [PubMed: 29420371]
- Wong BKY, Sutton VR. Aicardi syndrome, an unsolved mystery: Review of diagnostic features, previous attempts, and future opportunities for genetic examination. Am J Med Genet C Semin Med Genet. 2018;178:423–31. [PubMed: 30536540]
- Yilmaz S, Fontaine H, Brochet K, Gregoire MJ, Devignes MD, Schaff JL, Philippe C, Nemos C, McGregor JL, Jonveaux P. Screening of subtle copy number changes in Aicardi syndrome patients with a high resolution X chromosome array-CGH. Eur J Med Genet. 2007;50:386–91. [PubMed: 17625997]
- Zubairi MS, Carter RF, Ronen GM. A male phenotype with Aicardi syndrome. J Child Neurol. 2009;24:204–7. [PubMed: 19182158]
Publication Details
Author Information and Affiliations
Baylor College of Medicine & Texas Children's Hospital
Houston, Texas
Baylor College of Medicine
Houston, Texas
Publication History
Initial Posting: June 30, 2006; Last Update: November 12, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Sutton VR, Van den Veyver IB. Aicardi Syndrome. 2006 Jun 30 [Updated 2020 Nov 12]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.