Summary
Clinical characteristics.
Most characteristically, Aicardi-Goutières syndrome (AGS) manifests as an early-onset encephalopathy that usually, but not always, results in severe intellectual and physical disability. A subgroup of infants with AGS present at birth with abnormal neurologic findings, hepatosplenomegaly, elevated liver enzymes, and thrombocytopenia, a picture highly suggestive of congenital infection. Otherwise, most affected infants present at variable times after the first few weeks of life, frequently after a period of apparently normal development. Typically, they demonstrate the subacute onset of a severe encephalopathy characterized by extreme irritability, intermittent sterile pyrexias, loss of skills, and slowing of head growth. Over time, as many as 40% develop chilblain skin lesions on the fingers, toes, and ears. It is becoming apparent that atypical, sometimes milder, cases of AGS exist, and thus the true extent of the phenotype associated with pathogenic variants in the AGS-related genes is not yet known.
Diagnosis/testing.
The diagnosis of AGS is established in a proband with typical clinical findings and characteristic abnormalities on cranial CT (calcification of the basal ganglia and white matter) and MRI (leukodystrophic changes); AND/OR by identification of one of the following:
- Biallelic pathogenic variants in ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, or TREX1
- Specific heterozygous autosomal dominant pathogenic variants in TREX1 and ADAR
- A variety of heterozygous autosomal dominant pathogenic variants in IFIH1
Management.
Treatment of manifestations: Chest physiotherapy and treatment of respiratory complications; attention to diet and feeding methods to assure adequate caloric intake and avoid aspiration; management of seizures using standard protocols.
Surveillance: Monitoring for signs of diabetes insipidus in the neonatal period; repeat ophthalmologic examinations at least for the first few years of life to evaluate for evidence of glaucoma; monitoring for evidence of scoliosis, insulin-dependent diabetes mellitus, and hypothyroidism.
Genetic counseling.
AGS is most frequently inherited in an autosomal recessive manner; in a few instances the disease can result from specific de novo or inherited autosomal dominant pathogenic variants in ADAR or TREX1, and a variety of heterozygous autosomal dominant pathogenic variants in IFIH1. At conception, each sib of an affected individual with autosomal recessive AGS has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Individuals with AGS do not typically reproduce. Once the pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk for AGS, and preimplantation genetic testing are possible.
Diagnosis
In its most characteristic form, Aicardi-Goutières syndrome (AGS) can be considered an early-onset encephalopathy associated with significant intellectual and physical disability.
Suggestive Findings
Aicardi-Goutières syndrome (AGS) should be suspected in individuals with the following clinical, neuroimaging, and supportive laboratory findings [Goutières et al 1998, Lanzi et al 2002, Rice et al 2007b, Livingston et al 2013].
Clinical features
- Encephalopathy and/or significant intellectual disability
- Acquired microcephaly during the first year of life
- Dystonia and spasticity
- Sterile pyrexias
- Hepatosplenomegaly
- Chilblain lesions on the feet, hands, ears, and sometimes more generalized mottling of the skin. See Figure 1.
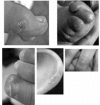
Figure 1.
Examples of chilblains seen in AGS Rice et al [2007b]; reprinted with permission of The American Journal of Human Genetics, University of Chicago Press.
Exclusion criteria include the following:
- Evidence of prenatal/perinatal infection including, but not limited to, CMV, toxoplasmosis, rubella, herpes simplex, Zika, and HIV
- Evidence of a known other metabolic disorder or neurodegenerative disorder
Neuroimaging
- Calcification (best visualized on CT scan) of the basal ganglia, particularly the putamen, globus pallidus and thalamus but also extending into the white matter, sometimes in a para- (rather than true peri-) ventricular distribution [Lanzi et al 2002, Uggetti et al 2009, Livingston et al 2013]. See Figure 2.Note: Intracranial calcification is not always recognized on MRI, the initial imaging modality employed in most units.
- White matter changes, particularly affecting the frontotemporal regions with (in severe cases) temporal lobe cyst-like formation. See Figure 3.On MRI, appears on T2-weighted images as a hyperintense signal most commonly located around the horns of the ventricles (Figure 3B)
- Cerebral atrophy, which may be progressive and involve the periventricular white matter and sulciCerebellar atrophy and brain stem atrophy may also be prominent (Figure 3D) [Crow et al 2004a, Sanchis et al 2005].
- Bilateral striatal necrosis
- Intracerebral vasculopathy including intracranial stenosis, moyamoya, and aneurysms
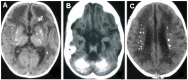
Figure 2.
Examples of intracranial calcification on CT scan in individuals with AGS. Calcification is seen: A. In the basal ganglia;
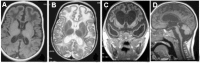
Figure 3.
The spectrum of brain changes seen on MRI in AGS A. Hypointensity on T1-weighted imaging of the white matter
Supportive Laboratory Findings
Peripheral blood
- Positive interferon signature identified using quantitative PCR analysis of RNA/cDNA [Rice et al 2013a, Crow et al 2015]
- Elevated liver enzymes
- Thrombocytopenia
Cerebrospinal fluid (CSF)
- Chronic CSF leukocytosis, defined as more than five lymphocytes/mm3 CSF.
- Typical values range from five to 100 lymphocytes/mm3 [Goutières et al 1998, Rice et al 2007b, Rice et al 2013a].
- A decrease in the number of lymphocytes occurs with time, although high cell counts may persist for several years.
- A normal cell count can be observed in the presence of elevated concentrations of IFN-α in the CSF even at an early stage of the disease [Crow et al 2003, Rice et al 2007b].
- Increased interferon-alpha (IFN-α) activity in the CSF (normal: <2 IU/mL)
- Recorded IFN-α activity is usually highest in the early stages of the disease. The IFN-α CSF activity can normalize over the first three to four years of life [Rice et al 2007b, Rice et al 2013a].
- Recorded IFN-α activity is usually higher in CSF than in blood, where it may be normal.
- High IFN-α activity has been identified in fetal blood at 26 weeks' gestation [Desanges et al 2006].
- Increased concentration of neopterin in the CSF [Rice et al 2007b]
- Levels are highest in the early stages of the disease and can normalize over time.
- Levels of the neurotransmitter metabolites 5HIAA, HVA, and 5MTHF are normal.
Establishing the Diagnosis
The diagnosis of AGS is established in a proband with typical clinical findings and characteristic abnormalities on cranial CT (calcification of the basal ganglia and white matter) and MRI (leukodystrophic changes) and/or by the identification of biallelic pathogenic (or likely pathogenic) variants in ADAR, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, or TREX1; specific heterozygous autosomal dominant pathogenic (or likely pathogenic) variants in TREX1 and ADAR; or a variety of heterozygous autosomal dominant pathogenic (or likely pathogenic) variants in IFIH1.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) The identification of variant(s) of uncertain significance cannot be used to confirm or rule out the diagnosis.
Molecular testing approaches can include serial single-gene testing, use of a multigene panel, and more comprehensive genomic testing.
Serial single-gene testing can be pursued based on the individual's ethnicity and/or in the order in which pathogenic variants most commonly occur (see Table 1).
- If only one pathogenic variant is identified in RNASEH2A, RNASEH2B, RNASEH2C, or SAMHD1, gene-targeted deletion/duplication analysis can be considered next.
- Gene-targeted deletion/duplication analysis may also be considered if a heterozygous pathogenic variant that is not known to be associated with autosomal dominant AGS is identified in TREX1 or ADAR.
A multigene panel that includes ADAR, IFIH1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1, and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included and the sensitivity of multigene panels vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
More comprehensive genomic testing (when available) including exome sequencing, mitochondrial sequencing, and genome sequencing may be considered if serial single-gene testing (and/or use of a multigene panel that includes ADAR, IFIH1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and TREX1) fails to confirm a diagnosis in an individual with features of AGS. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation). For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Aicardi-Goutières Syndrome
Clinical Characteristics
Clinical Description
In its most characteristic form, Aicardi-Goutières syndrome (AGS) can be considered an early-onset encephalopathy associated with significant intellectual and physical disability.
Pregnancy, delivery, and the neonatal period are normal in approximately 80% of infants with Aicardi-Goutières syndrome (AGS) [Rice et al 2007b]. However, brain calcifications can be identified in utero [Le Garrec et al 2005] and 20% of cases, mainly those caused by biallelic pathogenic variants in TREX1, present at birth with abnormal neurologic findings, hepatosplenomegaly, elevated liver enzymes, and thrombocytopenia, a picture reminiscent of congenital infection.
All other affected infants present at variable times after the first few weeks of life, frequently after a period of apparently normal development. The majority of these later-presenting infants exhibit subacute onset of a severe encephalopathy characterized by extreme irritability, intermittent sterile pyrexias, loss of skills, and slowing of head growth. The encephalopathic phase usually lasts several months. The opinion of most pediatricians caring for such children is that the disease does not progress beyond the encephalopathic period; occasionally, however, affected individuals do appear to show progression and/or episodes of regression. Death is usually considered to be secondary to the neurologic damage incurred during the initial disease episode, not to further regression. Several affected individuals older than age 30 years show no obvious signs of disease progression.
Neurologic features. Typically, affected individuals have peripheral spasticity, dystonic posturing (particularly of the upper limbs), truncal hypotonia, and poor head control. Seizures are reported in up to half of affected children, but are usually relatively easily controlled [Goutières et al 1998, Rice et al 2007b]. A number of children demonstrate a marked startle reaction to sudden noise, and the differentiation from epilepsy can be difficult. Most affected individuals have severe intellectual and physical impairment. Variability in the severity of the neurologic outcome can be observed among sibs. Most affected children exhibit a severe acquired microcephaly; in children with preserved intellect head circumference can be normal.
Hearing is almost always normal.
Ophthalmology. Visual function varies from normal to cortical blindness. Ocular structures are almost invariably normal on examination. However, there is a risk of congenital glaucoma or later-onset glaucoma [Crow et al 2004a, Crow et al 2015].
Skin lesions. As many as 40% of affected individuals [Rice et al 2007b] have skin lesions with chilblains on the fingers and toes and sometimes the ears and other pressure points (e.g., elbows) [Tolmie et al 1995, Stephenson 2002] (see Figure 1). The cutaneous lesions may be complicated by periungual infection and necrosis.
Other
- Intracranial large-vessel disease. An additional previously undescribed feature of AGS, which so far appears to be almost exclusively related to biallelic pathogenic variants in SAMHD1, is intracranial large-vessel disease causing both intracranial stenoses (in some cases reminiscent of moyamoya disease) and aneurysms (see Phenotype Correlations by Gene) [Ramesh et al 2010, Thiele et al 2010, du Moulin et al 2011, Xin et al 2011].
- Refractory four-limb dystonia. Several individuals with ADAR pathogenic variants have been reported to demonstrate an acute or subacute onset of refractory four-limb dystonia starting between age eight months and five years [Livingston et al 2014a, Crow et al 2015] (see Phenotype Correlations by Gene).
- Individuals can be developmentally normal at initial presentation.
- This phenotype can occur due to either biallelic pathogenic variants in ADAR or the autosomal dominant pathogenic variant p.Gly1007Arg in ADAR.
- Like other AGS-related phenotypes, bilateral striatal necrosis due to biallelic pathogenic variants in ADAR or the recurrent dominant pathogenic p.Gly1007Arg variant in ADAR are typically associated with an upregulation of ISGs.
Additional infrequently observed features of AGS are summarized in Table 2.
Table 2.
Infrequent Features Seen in a Cohort of 123 Individuals with Molecularly Confirmed AGS
Neuroimaging/neuropathologic findings
- Calcifications on neuroimaging:
- Are often punctate but may be dense and rock-like;
- When identified at diagnosis tend to remain stable, although progression can be observed [Lanzi et al 2002, Lanzi et al 2005];
- Are not correlated with the severity of neurologic outcome; in rare cases, calcifications may be absent and may or may not be seen on repeat scans. Therefore, their absence does not rule out the diagnosis [Aicardi & Goutières 1984].
- The main neuropathologic findings identified in severely affected individuals include [Kumar et al 1998, Barth 2002]:
- Diffuse but nonhomogeneous demyelination with astrocytosis; absence of signs of storage or myelin breakdown;
- Multiple wedge-shaped microinfarcts in the neocortex and cerebellar cortex, suggestive of a microangiopathy;
- Calcific deposits in the white matter, thalami, basal ganglia, and dentate nuclei;
- Calcification in the media, adventitia, and perivascular spaces of small vessels;
- Inflammation in the leptomeninges and areas of necrosis.
Phenotype Correlations by Gene
In general, the early-onset neonatal form of AGS is most frequently seen in association with biallelic pathogenic variants in RNASEH2A, RNASEH2C, or TREX1. The later-onset presentation (sometimes occurring after several months of normal development and occasionally associated with remarkably preserved neurologic function) is most frequently seen in association with biallelic pathogenic variants in RNASEH2B, SAMHD1, or ADAR but may also be seen in individuals who have an autosomal dominant heterozygous pathogenic variant in ADAR or IFIH1 [Crow et al 2015].
Mortality is correlated with genotype: 34% of individuals with RNASEH2A, RNASEH2C, and TREX1 pathogenic variants were known to have died compared to 8% with RNASEH2B pathogenic variants (p=0.001) [Rice et al 2007b]. The mortality associated with other genotypes is less clear.
ADAR. Pathogenic variants in ADAR have been associated with a clinical presentation of acute bilateral striatal necrosis.
A subgroup of individuals with ADAR pathogenic variants can present with symmetric signal changes in the caudate and putamen, often associated with swelling and later shrinkage in the context of an acute or subacute onset of refractory four-limb dystonia. Cases have been identified with onset as late as age four years on a background of completely normal development [La Piana et al 2014, Livingston et al 2014a].
ADAR-related disease should be considered in the differential diagnosis of apparently nonsyndromic bilateral striatal necrosis (BSN) with severe dystonia of varying evolution. The finding of an interferon signature provides a useful screening test for the presence of ADAR pathogenic variants in this context.
RNASEH2B. Some individuals with biallelic pathogenic variants in RNASEH2B have relatively preserved intellectual function, with a few having a completely normal IQ and head circumference [McEntagart et al 1998].
RNASEH2C. A family with a recurrent Asian founder variant in RNASEH2C and striking intrafamilial variability has been described [Crow et al 2015].
SAMHD1
- Intracerebral vasculopathy, including intracranial stenosis and aneurysms, is observed more frequently in individuals who have biallelic pathogenic variants in SAMHD1.
- Dale et al [2010] reported two sibs who had biallelic null variants in SAMHD1. The older girl showed mild intellectual disability with microcephaly. Her younger brother had significant spastic paraparesis with normal intellect and head size. Both children had an unclassified chronic inflammatory skin condition with chilblains and recurrent mouth ulcers. One of the sibs had a chronic progressive deforming arthropathy of the small and large joints with secondary contractures. Similar joint involvement was described by Ramantani et al [2010] (see also Crow et al [2015]).
- Biallelic pathogenic variants in SAMHD1 are most frequently associated with chilblains and glaucoma [Crow et al 2015].
Nomenclature
The microcephaly-intracranial calcification syndrome (MICS; also known as pseudo-TORCH syndrome or Baraitser-Reardon syndrome) was previously differentiated from AGS on the basis of congenital microcephaly and the presence of non-neurologic abnormalities including elevation of liver enzymes, hepatomegaly, and thrombocytopenia at birth [Reardon et al 1994]. However, recent studies have shown that these same features can be seen in persons with AGS in whom pathogenic variants in one of the associated genes have been identified [Rice et al 2007b]. Of note, in the majority of MICS cases reported no information is available on CSF cell count and IFN-α concentration; thus it is probable that most cases of MICS are in fact AGS.
"Familial systemic lupus erythematosus." Dale et al [2000] described two children of consanguineous parents with early-onset encephalopathy, intracranial calcifications, chilblain skin lesions, and the progressive production of high levels of autoantibodies. CSF was not analyzed. These cases most likely represent AGS [Aicardi & Goutières 2000]. Additional individuals with AGS who have signs and symptoms similar to systemic lupus erythematosus have also been described.
Prevalence
The actual frequency of AGS is unknown.
Pathogenic variants have been found in affected individuals of all ethnic origins [Crow et al 2006a, Crow et al 2006b, Rice et al 2007b, Rice et al 2013b] (see Table 1).
Genetically Related (Allelic) Disorders
Other phenotypes caused by pathogenic variants in ADAR, IFIH1, RNASEH2B, and TREX1 are listed in Table 3.
Table 3.
Allelic Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with pathogenic variants in RNASEH2A or RNASEH2C.
Differential Diagnosis
Calcification of the basal ganglia is a nonspecific finding seen in many diseases. However, in the context of an early-onset encephalopathy, conditions to consider include the following:
- TORCH congenital infections are the most common conditions in the differential and the most important to rule out because misdiagnosis would result in erroneous counseling as to risk of recurrence.Note: Other congenital infections, such as those associated with Zika and HIV, should also be considered in the differential diagnosis.
- The microcephaly-intracranial calcification syndrome (MICS). Given the phenotype of early-onset Aicardi-Goutières syndrome (AGS) cases (see Clinical Characteristics) [Reardon et al 1994], it is likely that most cases of MICS are in fact AGS (see Nomenclature). However, a number of other phenotypes are associated with neonatal intracranial calcification [Knoblauch et al 2003, Gardner et al 2005]; thus, this phenotype undoubtedly represents a heterogeneous group of diseases (see Nomenclature).
- Band-like calcification polymicrogyria (BLC-PMG; pseudo-TORCH syndrome) (OMIM 251290) [Abdel-Salam et al 2008, Briggs et al 2008] shows radiologic and clinical overlap with AGS, demonstrating intracranial calcification and significant psychomotor retardation with microcephaly and epilepsy. The condition can be differentiated by the observation of polymicrogyria (which has never been reported in AGS) and the identification of biallelic pathogenic variants in OCLN [O'Driscoll et al 2010].
- Superficially, at least, the MRI scan findings in AGS with frontotemporal white matter changes and cysts can be confused with Alexander disease, megalencephalic leukoencephalopathy with subcortical cysts, and childhood ataxia with central nervous system hypomyelination/vanishing white matter disease. The degree of white matter hypomyelination at an early age has also prompted consideration of Pelizeaus-Merzbacher disease in some individuals. In general terms, AGS should be considered in the differential diagnosis of an unexplained leukoencephalopathy. This clinical point is of particular importance because intracranial calcification is not always recognized on MRI, the initial imaging modality employed in most medical facilities.
- Classic Cockayne syndrome (CS type 1), a leukodystrophy with striocerebellar calcifications, is variably characterized by its distinctive facial features, dwarfism, nerve deafness, cataracts, retinal dystrophy, and skin photosensitivity. Inheritance is autosomal recessive.
- Neonatal lupus erythematosus. Prendiville et al [2003] described basal ganglia calcifications and patchy white matter attenuation in infants with neonatal lupus erythematosus reminiscent of the imaging findings seen in AGS. These children demonstrated extensive erythematous skin lesions distinct from the chilblain lesions seen in AGS. The authors reported normal neurologic outcome in these cases.
- Hoyeraal Hreidarsson syndrome, a severe form of dyskeratosis congenita, is caused by mutation in DKC1 (X-linked) or TINF2 (autosomal dominant). Hoyeraal Hreidarsson syndrome presents in the first months of life with microcephaly, cerebellar hypoplasia, and intracerebral calcifications. Affected males develop a pancytopenia that persists (in contrast to the thrombocytopenia seen in some individuals with AGS, which usually resolves in the first few weeks of life).
- Mitochondrial cytopathies, including Leigh syndrome and the familial mitochondrial encephalopathy with intracerebral calcifications described by Samson et al [1994]. See also Mitochondrial Disorders Overview.
- 3-hydroxyisobutyric aciduria (OMIM 236795). Chitayat et al [1992] described monozygotic male twins, born to nonconsanguineous parents, who had dysmorphic facial features, microcephaly, migrational brain disorder, and congenital intracerebral calcification.
- Blau et al [2003] described three individuals with microcephaly, severe intellectual disability and motor retardation, dyskinesia, spasticity, and occasional seizures with extremely high CSF concentrations of neopterin and biopterin and low CSF concentration of 5-methyltetrahydrofolate. Although reported as having AGS, they did not demonstrate a CSF lymphocytosis or elevation of IFN-α concentration. Thus, these individuals may have an undefined syndrome within the group of infants with encephalopathy and intracranial calcifications. However, it is now known that a similar pterin profile can be observed in individuals with molecularly confirmed AGS [Rice et al 2007b].
- Cerebroretinal microangiopathy with calcifications and cysts (CRMCC; Coats plus) (OMIM 612199) is caused by biallelic pathogenic variants in CTC1 [Crow et al 2004b, Linnankivi et al 2006, Anderson et al 2012, Livingston et al 2014b].
- Leukoencephalopathy, brain calcifications, and cysts (Labrune syndrome) (OMIM 614561)
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Aicardi-Goutières syndrome (AGS), the following evaluations are recommended:
- Developmental assessment
- Assessment of feeding and nutritional status
- Ophthalmologic examination
- EEG to evaluate for seizures, if suspected
- Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
The following are appropriate:
- Chest physiotherapy and vigorous treatment of respiratory complications
- Attention to diet and method of feeding to assure adequate caloric intake
- Management of seizures using standard protocols
Surveillance
Surveillance includes the following:
- Monitoring for signs of diabetes insipidus in the neonatal period
- Assessment for glaucoma at least for the first few years of life
- Monitoring of the spine for the development of scoliosis
- Monitoring for signs of insulin-dependent diabetes mellitus and hypothyroidism
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Research into the role of immunosuppressive agents in the treatment of AGS is ongoing [Crow & Rehwinkel 2009, Crow et al 2014].
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Other
Corticosteroids can lower the CSF concentration of interferon [PG Barth 2003, personal communication]; the clinical benefit of such treatment is unproven.
Note: The description of intracranial large-vessel disease in association with biallelic pathogenic variants in SAMHD1 raises important questions about the management of such individuals. The occlusive and aneurysmal arteriopathies described could be amenable to treatment (revascularization for the former and coiling or clipping for the latter). Moreover, the likely inflammatory basis of the arteriopathy suggests that immunosuppression may play a role in management. A key question is whether inflammatory disease is active at the time of clinical presentation, or whether the arterial abnormalities observed represent the end result of a now-quiescent inflammatory process.
Given the lack of evidence, no definitive statement about these issues can be made at present. However, the potential for intervention exists, and it could be argued that some individuals (e.g., those with lesser psychomotor problems) warrant such intervention and should be actively screened for intracranial arteriopathy, if only by close inspection of the vasculature at the base of the brain seen on routine MRI. Chilblains were present in all the affected individuals described by Ramesh et al [2010], and it may be that their presence predicts an increased risk for intracranial vasculopathy.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
RNASEH2A, RNASEH2B, RNASEH2C, and SAMHD1-related Aicardi-Goutières syndrome (AGS) is inherited in an autosomal recessive manner.
TREX1 and ADAR-related AGS can be inherited in an autosomal recessive or autosomal dominant manner depending on the specific pathogenic variant.
IFIH1-related Aicardi-Goutières syndrome (AGS) is inherited in an autosomal dominant manner.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., carriers of one AGS-related pathogenic variant).
- Heterozygotes (carriers) are not at risk of developing AGS; however, the findings of Lee-Kirsch et al [2007b] and Richards et al [2007] suggest that heterozygotes may be at increased risk of developing later-onset systemic lupus erythematosus (SLE) or retinal vasculopathy with cerebral leukodystrophy (RVCL), depending on the specific gene and pathogenic variant involved (see Genetically Related Disorders).
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are not at risk of developing AGS; however, heterozygotes may be at increased risk of developing later-onset systemic lupus erythematosus (SLE) or retinal vasculopathy with cerebral leukodystrophy (RVCL), depending on the specific gene and pathogenic variant involved (see Genetically Related Disorders).
Offspring of a proband. Most individuals with AGS do not reproduce.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an AGS-related pathogenic variant.
Carrier (heterozygote) detection. Carrier testing for at-risk relatives requires prior identification of the AGS-related pathogenic variants in the family.
Autosomal Dominant Inheritance – Risk to Family Members
Parents of a proband
- To date, most probands with autosomal dominant AGS have had the disorder as a result of a de novo TREX1, ADAR, or IFIH1 pathogenic variant [Rice et al 2007b, Haaxma et al 2010, Rice et al 2012, Abe et al 2014].Vertical transmission of pathogenic variants in TREX1 (p.Asp18Asn; Abe et al [2013]) and ADAR (p.Gly1007Arg; Rice et al [2012], Livingston et al [2014a]) has been reported.
- Molecular genetic testing is recommended for the evaluation of parents of a proband with an apparent de novo pathogenic variant.
- If the pathogenic variant found in the proband cannot be detected in leukocyte DNA of either parent, the proband most likely has a de novo pathogenic variant. Another possible explanation is germline mosaicism in a parent (though theoretically possible, no instances of germline mosaicism have been reported).
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband is affected, the risk to the sibs is 50%.
- If the TREX1, ADAR, or IFIH1 pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the risk to sibs is presumed to be slightly greater than that of the general population (though still <1%) because of the theoretic possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with autosomal dominant AGS has a 50% chance of inheriting the AGS-related pathogenic variant; however, most individuals with AGS do not reproduce.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected, the parent's family members may be at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, allelic variants, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative genetic alteration/s are unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Once the pathogenic variant(s) have been identified in an affected family member, prenatal and preimplantation genetic testing for AGS are possible.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- International Aicardi-Goutières Syndrome Association (IAGSA)ItalyEmail: associazione.iagsa@tiscali.it; iagsa@libero.it
- MedlinePlus
- United Leukodystrophy Foundation (ULF)224 North Second StreetSuite 2DeKalb IL 60115Phone: 800-728-5483 (toll-free); 815-748-3211Fax: 815-748-0844Email: office@ulf.org
- Myelin Disorders Bioregistry ProjectPhone: 215-590-1719Email: sherbinio@chop.edu
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Aicardi-Goutieres Syndrome: Genes and Databases
Table B.
OMIM Entries for Aicardi-Goutieres Syndrome (View All in OMIM)
ADAR
Gene structure. ADAR is a single-copy 16-exon gene that encodes two main isoforms constitutively expressed in mammalian cells: a truncated protein (p110 NP_001020278.1) encoded by a transcript variant of 15 exons (NM_001025107.2) and an IFN inducible full-length protein (p150 NP_001102.2) induced isoform encoded by transcript variant NM_001111.4. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. The majority of pathogenic variants in ADAR result in autosomal recessive disease. Missense, nonsense, frameshift, and splice site pathogenic variants have been reported. One missense variant, p.Pro193Ala, is common in individuals of European origin. One dominant variant, p.Gly1007Arg, has been reported [Rice et al 2012, Livingston et al 2014a].
Table 4.
Selected ADAR Pathogenic Variants
Normal gene product. DRADA, double-stranded RNA-specific adenosine deaminase, is a modular protein with a C-terminal deaminase catalytic domain, three centrally located dsRNA-binding domains (dsRBDs) and one or two N-terminal Z-DNA–binding domains; compared with p110, the p150 isoform of human DRADA possesses an additional 295 N-terminal amino acids containing a nuclear export signal and an extra Z-DNA/Z-RNA-binding domain.
Abnormal gene product. Pathogenic variants in ADAR are believed to cause AGS as a result of loss of protein activity; the precise mechanism leading to disease is unclear.
IFIH1
Gene structure. IFIH1 has 16 exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. All IFIH1 pathogenic variants associated with AGS to date have been missense variants that cause dominant disease.
Normal gene product. IFIH1 encodes the interferon-induced helicase C domain-containing protein 1, a cytosolic double-stranded RNA receptor.
Abnormal gene product. Pathogenic variants in IHIF1 associated with AGS encode missense variants that cause up-regulation of type I interferon signaling [Rice et al 2014].
RNASEH2A
Gene structure. RNASEH2A has eight exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. The majority of pathogenic variants in RNASEH2A are missense; splicing and frameshift pathogenic variants have also been reported [Crow et al 2006b, Rice et al 2007b] (see Table 5).
Table 5.
Selected RNASEH2A Pathogenic Variants
Normal gene product. RNASEH2A encodes the ribonuclease H2 subunit A, which comprises 299 amino acids. Ribonuclease H (RNASEH) enzymes endonucleolytically cleave ribonucleotides from RNA:DNA duplexes. RNASEH2 has been proposed to function in the removal of lagging strand Okazaki fragment RNA primers during DNA replication, as well as in the excision of single ribonucleotides from DNA:DNA duplexes. However, the precise biologic function of the human RNASEH2 complex in the context of AGS is uncertain.
Abnormal gene product. See RNASEH2B, Abnormal gene product.
RNASEH2B
Gene structure. RNASEH2B has 11 exons and codes for a 308-amino acid protein. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. Almost all pathogenic variants so far identified in RNASEH2B are missense [Crow et al 2006b, Rice et al 2007b] (see Table 6).
The frequency of alleles identified in affected individuals is:
- p.Ala177Thr (62%)
- p.Thr163Ile (7%)
- p.Val185Gly (7%)
- c.136+1delG (4%)
- Remaining alleles (<2%) [Rice et al 2007b]
Table 6.
Selected RNASEH2B Pathogenic Variants
Normal gene product. The precise function of the ribonuclease H2 subunit B protein within the human RNASEH2 complex is unknown.
Abnormal gene product. Pathogenic variants in genes encoding any of the three subunits of the ribonuclease H2 complex are thought to cause AGS resulting from a loss of enzymatic function.
RNASEH2C
Gene structure. RNASEH2C is a four-exon gene encoding a 164-amino acid protein. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. All pathogenic variants so far identified in RNASEH2C are missense [Crow et al 2006b, Rice et al 2007b] (see Table 7).
The frequency of alleles identified in affected individuals is:
- p.Arg69Trp (72%)
- Remaining alleles (seen only in single families)
Table 7.
Selected RNASEH2C Pathogenic Variants
Normal gene product. The function of ribonuclease H2 subunit C (RNASEH2C) within the RNASEH2 complex is unknown.
Abnormal gene product. See RNASEH2B, Abnormal gene product.
SAMHD1
Gene structure. SAMHD1 has 16 exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. The majority of pathogenic variants are missense, splice site, nonsense, or frameshift variants [Rice et al 2009].
Three large deletions plus a small deletion have also been identified. A large deletion of exon 1 is common among individuals of Ashkenazi Jewish descent [Crow et al 2015].
Table 8.
Selected SAMHD1 Pathogenic Variants
Normal gene product. SAMHD1 is a 626-amino acid protein that consists of a SAM domain and an HD domain; the protein acts as a dNTP triphosphohydrolase [Goldstone et al 2011].
Abnormal gene product. Pathogenic variants in SAMHD1 are believed to cause AGS as a result of mislocalization of the protein or loss of protein function.
TREX1 (AGS1)
Gene structure. TREX1 has one exon. For a detailed summary of gene and protein information, see Table A, Gene.
Note: A great deal of confusion regarding TREX1 and the overlapping gene ATRIP exists in the databases. TREX1 and ATRIP are distinct genes that encode distinct proteins; they are not known to be relevant to one another [Yang et al 2007].
Pathogenic variants. Stop variants, deletions, and insertions are common in TREX1, but the most prevalent pathogenic variant is a missense variant (p.Arg114His) that affects the dimerization of the TREX1 protein (3' repair exonuclease 1) and is likely to be a functional null allele. The pathogenic variant p.Arg114His is particularly common in people from northern Europe.
Affected individuals are almost always homozygotes or compound heterozygotes for pathogenic variants within the same gene. However, children with clinically typical AGS had a de novo heterozygous pathogenic variant in TREX1 [Rice et al 2007a, Haaxma et al 2010, Abe et al 2014] (see Table 9).
The frequency of alleles identified in affected individuals is [Rice et al 2007b]:
- p.Arg114His (50%)
- c.58_59insG (1%)
- Remaining alleles (<1%)
Table 9.
Selected TREX1 Pathogenic Variants
Normal gene product. TREX1 is a single-exon gene encoding a 314-amino acid residue protein.
TREX1 protein represents the major 3'→5' DNA exonuclease activity measured in mammalian cells. The protein has three conserved sequence motifs known as Exo I, II, and III. These motifs contain four conserved acidic residues that participate in coordination of divalent metal ions required for catalysis. In addition, the protein contains a C-terminal domain of about 75 amino acids, which is probably involved in subcellular localization of the protein, and a polyproline motif that may be involved in the interaction with other proteins. TREX1 appears to play a role in the disposal of single-stranded DNA possibly produced as a normal replication intermediate during S phase [Yang et al 2007], or derived from retro-elements [Stetson et al 2008].
Abnormal gene product. The most prevalent pathogenic variant in TREX1 is a missense change (p.Arg114His) that affects the dimerization of the TREX1 protein (3' repair exonuclease 1) and is likely to be a functional null allele, as are other reported frameshift variants.
Chapter Notes
Acknowledgments
We would like to thank Dr Gillian Rice for her help in compiling the gene variant data.
Author History
Jean Aicardi, MD, FRCP; Hospital Robert-Debré, Paris (2005-2014)
Yanick J Crow, MBBS, BMedSci, MRCP, PhD (2005-present)
John BP Stephenson, DM, FRCP, HonFRCPCH; Royal Hospital for Sick Children, Glasgow (2008-2014)
Revision History
- 22 November 2016 (ma) Comprehensive update posted live
- 13 March 2014 (me) Comprehensive update posted live
- 1 March 2012 (cd) Revision: targeted mutation analysis for the c.490C>T mutation in TREX1 available clinically
- 19 January 2012 (cd) Revision: deletion/duplication analysis available clinically for SAMHD1, TREX1, RNASEH2A, and RNASEH2B; multigene panels available
- 4 November 2010 (me) Comprehensive update posted live
- 17 April 2008 (me) Comprehensive update posted live
- 29 June 2005 (me) Review posted live
- 1 September 2004 (ja) Original submission
References
Literature Cited
- Abdel-Salam GM, Zaki MS, Saleem SN, Gaber KR. Microcephaly, malformation of brain development and intracranial calcification in sibs: pseudo-TORCH or a new syndrome. Am J Med Genet A. 2008;146A:2929–36. [PubMed: 18925673]
- Abe J, Izawa K, Nishikomori R, Awaya T, Kawai T, Yasumi T, Hiragi N, Hiragi T, Ohshima Y, Heike T. Heterozygous TREX1 p.Asp18Asn mutation can cause variable neurological symptoms in a family with Aicardi-Goutieres syndrome/familial chilblain lupus. Rheumatology (Oxford). 2013;52:406–8. [PubMed: 22829693]
- Abe J, Nakamura K, Nishikomori R, Kato M, Mitsuiki N, Izawa K, Awaya T, Kawai T, Yasumi T, Toyoshima I, Hasegawa K, Ohshima Y, Hiragi T, Sasahara Y, Suzuki Y, Kikuchi M, Osaka H, Ohya T, Ninomiya S, Fujikawa S, Akasaka M, Iwata N, Kawakita A, Funatsuka M, Shintaku H, Ohara O, Ichinose H, Heike T. A nationwide survey of Aicardi-Goutieres syndrome patients identifies a strong association between dominant TREX1 mutations and chilblain lesions: Japanese cohort study. Rheumatology (Oxford). 2014;53:448–58. [PubMed: 24300241]
- Aicardi J, Goutières F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. 1984;15:49–54. [PubMed: 6712192]
- Aicardi J, Goutières F. Systemic lupus erythematosus or Aicardi-Goutieres syndrome? Neuropediatrics. 2000;31:113. [PubMed: 10963096]
- Anderson BH, Kasher PR, Mayer J, Szynkiewicz M, Jenkinson EM, Bhaskar SS, Urquhart JE, Daly SB, Dickerson JE, O'Sullivan J, Leibundgut EO, Muter J, Abdel-Salem GM, Babul-Hirji R, Baxter P, Berger A, Bonafé L, Brunstom-Hernandez JE, Buckard JA, Chitayat D, Chong WK, Cordelli DM, Ferreira P, Fluss J, Forrest EH, Franzoni E, Garone C, Hammans SR, Houge G, Hughes I, Jacquemont S, Jeannet PY, Jefferson RJ, Kumar R, Kutschke G, Lundberg S, Lourenço CM, Mehta R, Naidu S, Nischal KK, Nunes L, Ounap K, Philippart M, Prabhakar P, Risen SR, Schiffmann R, Soh C, Stephenson JB, Stewart H, Stone J, Tolmie JL, van der Knaap MS, Vieira JP, Vilain CN, Wakeling EL, Wermenbol V, Whitney A, Lovell SC, Meyer S, Livingston JH, Baerlocher GM, Black GC, Rice GI, Crow YJ. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat Genet. 2012;44:338–42. [PubMed: 22267198]
- Barth PG (2002) The neuropathology of Aicardi-Goutieres syndrome. Eur J Paediatr Neurol. 6 Suppl A:A27-31. [PubMed: 12365358]
- Blau N, Bonafe L, Krageloh-Mann I, Thony B, Kierat L, Hausler M, Ramaekers V. Cerebrospinal fluid pterins and folates in Aicardi-Goutieres syndrome: a new phenotype. Neurology. 2003;61:642–7. [PubMed: 12963755]
- Briggs TA, Wolf NI, D'Arrigo S, Ebinger F, Harting I, Dobyns WB, Livingston JH, Rice GI, Crooks D, Rowland-Hill CA, Squier W, Stoodley N, Pilz DT, Crow YJ. Band-like intracranial calcification with simplified gyration and polymicrogyria: a distinct "pseudo-TORCH" phenotype. Am J Med Genet A. 2008;146A:3173–80. [PubMed: 19012351]
- Chitayat D, Meagher-Villemure K, Mamer OA, O'Gorman A, Hoar DI, Silver K, Scriver CR. Brain dysgenesis and congenital intracerebral calcification associated with 3-hydroxyisobutyric aciduria. J Pediatr. 1992;121:86–9. [PubMed: 1625099]
- Crow YJ, Black DN, Ali M, Bond J, Jackson AP, Lefson M, Michaud J, Roberts E, Stephenson JB, Woods CG, Lebon P. Cree encephalitis is allelic with Aicardi-Goutieres syndrome: implications for the pathogenesis of disorders of interferon alpha metabolism. J Med Genet. 2003;40:183–7. [PMC free article: PMC1735395] [PubMed: 12624136]
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, Abdel-Hamid MS, Abdel-Salam GM, Ackroyd S, Aeby A, Agosta G, Albin C, Allon-Shalev S, Arellano M, Ariaudo G, Aswani V, Babul-Hirji R, Baildam EM, Bahi-Buisson N, Bailey KM, Barnerias C, Barth M, Battini R, Beresford MW, Bernard G, Bianchi M, Billette de Villemeur T, Blair EM, Bloom M, Burlina AB, Carpanelli ML, Carvalho DR, Castro-Gago M, Cavallini A, Cereda C, Chandler KE, Chitayat DA, Collins AE, Sierra Corcoles C, Cordeiro NJ, Crichiutti G, Dabydeen L, Dale RC, D'Arrigo S, De Goede CG, De Laet C, De Waele LM, Denzler I, Desguerre I, Devriendt K, Di Rocco M, Fahey MC, Fazzi E, Ferrie CD, Figueiredo A, Gener B, Goizet C, Gowrinathan NR, Gowrishankar K, Hanrahan D, Isidor B, Kara B, Khan N, King MD, Kirk EP, Kumar R, Lagae L, Landrieu P, Lauffer H, Laugel V, La Piana R, Lim MJ, Lin JP, Linnankivi T, Mackay MT, Marom DR, Marques Lourenço C, McKee SA, Moroni I, Morton JE, Moutard ML, Murray K, Nabbout R, Nampoothiri S, Nunez-Enamorado N, Oades PJ, Olivieri I, Ostergaard JR, Pérez-Dueñas B, Prendiville JS, Ramesh V, Rasmussen M, Régal L, Ricci F, Rio M, Rodriguez D, Roubertie A, Salvatici E, Segers KA, Sinha GP, Soler D, Spiegel R, Stödberg TI, Straussberg R, Swoboda KJ, Suri M, Tacke U, Tan TY, te Water Naude J, Wee Teik K, Thomas MM, Till M, Tonduti D, Valente EM, Van Coster RN, van der Knaap MS, Vassallo G, Vijzelaar R, Vogt J, Wallace GB, Wassmer E, Webb HJ, Whitehouse WP, Whitney RN, Zaki MS, Zuberi SM, Livingston JH, Rozenberg F, Lebon P, Vanderver A, Orcesi S, Rice GI. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. 2015;2015;167A:296–312. [PMC free article: PMC4382202] [PubMed: 25604658]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006a;38:917–20. [PubMed: 16845398]
- Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Dery C, Fazzi E, Goizet C, King MD, Klepper J, Lacombe D, Lanzi G, Lyall H, Martinez-Frias ML, Mathieu M, McKeown C, Monier A, Oade Y, Quarrell OW, Rittey CD, Rogers RC, Sanchis A, Stephenson JB, Tacke U, Till M, Tolmie JL, Tomlin P, Voit T, Weschke B, Woods CG, Lebon P, Bonthron DT, Ponting CP, Jackson AP. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006b;38:910–6. [PubMed: 16845400]
- Crow YJ, Massey RF, Innes JR, Pairaudeau PW, Rowland Hill CA, Woods CG, Ali M, Livingston JH, Lebon P, Nischall K, McEntagart M, Hindocha N, Winter RM. Congenital glaucoma and brain stem atrophy as features of Aicardi-Goutieres syndrome. Am J Med Genet. 2004a;129A:303–7. [PubMed: 15326633]
- Crow YJ, McMenamin J, Haenggeli CA, Hadley DM, Tirupathi S, Treacy EP, Zuberi SM, Browne BH, Tolmie JL, Stephenson JB. Coats' plus: a progressive familial syndrome of bilateral Coats' disease, characteristic cerebral calcification, leukoencephalopathy, slow pre- and post-natal linear growth and defects of bone marrow and integument. Neuropediatrics. 2004b;35:10–9. [PubMed: 15002047]
- Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. 2009;18:R130–6. [PMC free article: PMC2758706] [PubMed: 19808788]
- Crow YJ, Vanderver A, Orcesi S, Kuijpers TW, Rice GI. Therapies in Aicardi-Goutières syndrome. Clin Exp Immunol. 2014;175:1–8. [PMC free article: PMC3898548] [PubMed: 23607857]
- Crow YJ, Zaki MS, Abdel-Hamid MS, Abdel-Salam G, Boespflug-Tanguy O, Cordeiro NJ, Gleeson JG, Gowrinathan NR, Laugel V, Renaldo F, Rodriguez D, Livingston JH, Rice GI. Mutations in ADAR1, IFIH1, and RNASEH2B presenting as spastic paraplegia. Neuropediatrics. 2014a;45:386–93. [PubMed: 25243380]
- Dale RC, Gornall H, Singh-Grewal D, Alcausin M, Rice GI, Crow YJ. Familial Aicardi-Goutières syndrome due to SAMHD1 mutations is associated with chronic arthropathy and contractures. Am J Med Genet A. 2010;152A:938–42. [PubMed: 20358604]
- Dale RC, Tang SP, Heckmatt JZ, Tatnall FM. Familial systemic lupus erythematosus and congenital infection-like syndrome. Neuropediatrics. 2000;31:155–8. [PubMed: 10963105]
- Desanges C, Lebon P, Bauman C, Vuillard E, Garel C, Cordesse A, Oury JF, Crow Y, Luton D. Elevated interferon-alpha in fetal blood in the prenatal diagnosis of Aicardi-Goutieres syndrome. Fetal Diagn Ther. 2006;21:153–5. [PubMed: 16354995]
- du Moulin M, Nürnberg P, Crow YJ, Rutsch F. Cerebral vasculopathy is a common feature in Aicardi-Goutieres syndrome associated with SAMHD1 mutations. Proc Natl Acad Sci U S A. 2011;108:E232. [PMC free article: PMC3127916] [PubMed: 21633013]
- Gardner RJ, Chow CW, Simpson I, Fink AM, Meagher SE, White SM. Severe fetal brain dysgenesis with focal calcification. Prenat Diagn. 2005;25:362–4. [PubMed: 15906425]
- Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, de Carvalho LP, Stoye JP, Crow YJ, Taylor IA, Webb M. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480:379–82. [PubMed: 22056990]
- Goutières F, Aicardi J, Barth PG, Lebon P. Aicardi-Goutieres syndrome: an update and results of interferon-alpha studies. Ann Neurol. 1998;44:900–7. [PubMed: 9851434]
- Haaxma CA, Crow YJ, van Steensel MA, Lammens MM, Rice GI, Verbeek MM, Willemsen MA. A de novo p.Asp18Asn mutation in TREX1 in a patient with Aicardi-Goutières syndrome. Am J Med Genet A. 2010;152A:2612–7. [PubMed: 20799324]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Knoblauch H, Tennstedt C, Brueck W, Hammer H, Vulliamy T, Dokal I, Lehmann R, Hanefeld F, Tinschert S. Two brothers with findings resembling congenital intrauterine infection-like syndrome (pseudo-TORCH syndrome). Am J Med Genet A. 2003;120A:261–5. [PubMed: 12833411]
- Kondo T, Suzuki T, Ito S, Kono M, Negoro T, Tomita Y. Dyschromatosis symmetrica hereditaria associated with neurological disorders. J Dermatol. 2008;35:662–6. [PubMed: 19017046]
- Kumar D, Rittey C, Cameron AH, Variend S. Recognizable inherited syndrome of progressive central nervous system degeneration and generalized intracranial calcification with overlapping phenotype of the syndrome of Aicardi and Goutieres. Am J Med Genet. 1998;75:508–15. [PubMed: 9489795]
- La Piana R, Uggetti C, Olivieri I, Tonduti D, Balottin U, Fazzi E, Orcesi S. Bilateral striatal necrosis in two subjects with Aicardi-Goutières syndrome due to mutations in ADAR1 (AGS6). Am J Med Genet A. 2014;164A:815–9. [PubMed: 24376015]
- Lanzi G, Fazzi E, D'Arrigo S. Aicardi-Goutieres syndrome: a description of 21 new cases and a comparison with the literature. Eur J Paediatr Neurol. 2002;6:A9–A22. [PubMed: 12365365]
- Lanzi G, Fazzi E, D'Arrigo S, Orcesi S, Maraucci I, Uggetti C, Bertini E, Lebon P. The natural history of Aicardi-Goutieres syndrome: follow-up of 11 Italian patients. Neurology. 2005;64:1621–4. [PubMed: 15883328]
- Le Garrec M, Doret M, Pasquier JC, Till M, Lebon P, Buenerd A, Escalon J, Gaucherand P. Prenatal diagnosis of Aicardi-Goutieres syndrome. Prenat Diagn. 2005;25:28–30. [PubMed: 15662687]
- Lee-Kirsch MA, Chowdhury D, Harvey S, Gong M, Senenko L, Engel K, Pfeiffer C, Hollis T, Gahr M, Perrino FW, Lieberman J, Hubner N. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med. 2007a;85:531–7. [PubMed: 17440703]
- Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hubner N. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007b;39:1065–7. [PubMed: 17660818]
- Linnankivi T, Valanne L, Paetau A, Alafuzoff I, Hakumaki JM, Kivela T, Lonnqvist T, Makitie O, Paakkonen L, Vainionpaa L, Vanninen R, Herva R, Pihko H. Cerebroretinal microangiopathy with calcifications and cysts. Neurology. 2006;67:1437–43. [PubMed: 16943371]
- Livingston JH, Lin JP, Dale RC, Gill D, Brogan P, Munnich A, Kurian MA, Gonzalez-Martinez V, De Goede CG, Falconer A, Forte G, Jenkinson EM, Kasher PR, Szynkiewicz M, Rice GI, Crow YJ. A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J Med Genet. 2014a;51:76–82. [PubMed: 24262145]
- Livingston JH, Mayer J, Jenkinson E, Kasher P, Stivaros S, Berger A, Cordelli DM, Ferreira P, Jefferson R, Kutschke G, Lundberg S, Ounap K, Prabhakar P, Soh C, Stewart H, Stone J, van der Knaap MS, van Esch H, van Mol C, Wakeling E, Whitney A, Rice GI, Crow YJ. Leukoencephalopathy with calcifications and cysts: a purely neurological disorder distinct from Coats plus. Neuropediatrics. 2014b;45:175–82. [PubMed: 24407470]
- Livingston JH, Stivaros S, van der Knaap MS, Crow YJ. Recognizable phenotypes associated with intracranial calcification. Dev Med Child Neurol. 2013;55:46–57. [PubMed: 23121296]
- McEntagart M, Kamel H, Lebon P, King MD. Aicardi-Goutieres syndrome: an expanding phenotype. Neuropediatrics. 1998;29:163–7. [PubMed: 9706629]
- O'Driscoll MC, Daly SB, Urquhart JE, Black GC, Pilz DT, Brockmann K, McEntagart M, Abdel-Salam G, Zaki M, Wolf NI, Ladda RL, Sell S, D'Arrigo S, Squier W, Dobyns WB, Livingston JH, Crow YJ. Recessive mutations in the gene encoding the tight junction protein occludin cause band-like calcification with simplified gyration and polymicrogyria. Am J Hum Genet. 2010;87:354–64. [PMC free article: PMC2933344] [PubMed: 20727516]
- Prendiville JS, Cabral DA, Poskitt KJ, Au S, Sargent MA. Central nervous system involvement in neonatal lupus erythematosus. Pediatr Dermatol. 2003;20:60–7. [PubMed: 12558850]
- Ramantani G, Kohlhase J, Hertzberg C, Innes AM, Engel K, Hunger S, Borozdin W, Mah JK, Ungerath K, Walkenhorst H, Richardt HH, Buckard J, Bevot A, Siegel C, von Stülpnagel C, Ikonomidou C, Thomas K, Proud V, Niemann F, Wieczorek D, Häusler M, Niggemann P, Baltaci V, Conrad K, Lebon P, Lee-Kirsch MA. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutières syndrome. Arthritis Rheum. 2010;62:1469–77. [PubMed: 20131292]
- Ramesh V, Bernardi B, Stafa A, Garone C, Franzoni E, Abinun M, Mitchell P, Mitra D, Friswell M, Nelson J, Shalev SA, Rice GI, Gornall H, Szynkiewicz M, Aymard F, Ganesan V, Prendiville J, Livingston JH, Crow YJ. Intracerebral large artery disease in Aicardi-Goutières syndrome implicates SAMHD1 in vascular homeostasis. Dev Med Child Neurol. 2010;52:725–32. [PubMed: 20653736]
- Ravenscroft JC, Suri M, Rice GI, Szynkiewicz M, Crow Y. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am J Med Genet A. 2011;155A:235–7. [PubMed: 21204240]
- Reardon W, Hockey A, Silberstein P, Kendall B, Farag TI, Swash M, Stevenson R, Baraitser M. Autosomal recessive congenital intrauterine infection-like syndrome of microcephaly, intracranial calcification, and CNS disease. Am J Med Genet. 1994;52:58–65. [PubMed: 7977464]
- Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O'Hara A, Herrick AL, Bowden AP, Perrino FW, Lindahl T, Barnes DE, Crow YJ. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007a;80:811–5. [PMC free article: PMC1852703] [PubMed: 17357087]
- Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B, Baxter P, Benko WS, Bergmann C, Bertini E, Biancheri R, Blair EM, Blau N, Bonthron DT, Briggs T, Brueton LA, Brunner HG, Burke CJ, Carr IM, Carvalho DR, Chandler KE, Christen HJ, Corry PC, Cowan FM, Cox H, D'Arrigo S, Dean J, De Laet C, De Praeter C, Dery C, Ferrie CD, Flintoff K, Frints SG, Garcia-Cazorla A, Gener B, Goizet C, Goutieres F, Green AJ, Guet A, Hamel BC, Hayward BE, Heiberg A, Hennekam RC, Husson M, Jackson AP, Jayatunga R, Jiang YH, Kant SG, Kao A, King MD, Kingston HM, Klepper J, van der Knaap MS, Kornberg AJ, Kotzot D, Kratzer W, Lacombe D, Lagae L, Landrieu PG, Lanzi G, Leitch A, Lim MJ, Livingston JH, Lourenco CM, Lyall EG, Lynch SA, Lyons MJ, Marom D, McClure JP, McWilliam R, Melancon SB, Mewasingh LD, Moutard ML, Nischal KK, Ostergaard JR, Prendiville J, Rasmussen M, Rogers RC, Roland D, Rosser EM, Rostasy K, Roubertie A, Sanchis A, Schiffmann R, Scholl-Burgi S, Seal S, Shalev SA, Corcoles CS, Sinha GP, Soler D, Spiegel R, Stephenson JB, Tacke U, Tan TY, Till M, Tolmie JL, Tomlin P, Vagnarelli F, Valente EM, Van Coster RN, Van der Aa N, Vanderver A, Vles JS, Voit T, Wassmer E, Weschke B, Whiteford ML, Willemsen MA, Zankl A, Zuberi SM, Orcesi S, Fazzi E, Lebon P, Crow YJ. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. 2007b;81:713–25. [PMC free article: PMC2227922] [PubMed: 17846997]
- Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, Couthard LR, Aeby A, Attard-Montalto SP, Bertini E, Bodemer C, Brockmann K, Brueton LA, Corry PC, Desguerre I, Fazzi E, Cazorla AG, Gener B, Hamel BC, Heiberg A, Hunter M, van der Knaap MS, Kumar R, Lagae L, Landrieu PG, Lourenco CM, Marom D, McDermott MF, van der Merwe W, Orcesi S, Prendiville JS, Rasmussen M, Shalev SA, Soler DM, Shinawi M, Spiegel R, Tan TY, Vanderver A, Wakeling EL, Wassmer E, Whittaker E, Lebon P, Stetson DB, Bonthron DT, Crow YJ. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41:829–32. [PMC free article: PMC4154505] [PubMed: 19525956]
- Rice GI, del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, Bader-Meunier B, Baildam EM, Battini R, Beresford MW, Casarano M, Chouchane M, Cimaz R, Collins AE, Cordeiro NJ, Dale RC, Davidson JE, De Waele L, Desguerre I, Faivre L, Fazzi E, Isidor B, Lagae L, Latchman AR. Lebon P2, Li C, Livingston JH, Lourenço CM, Mancardi MM, Masurel-Paulet A, McInnes IB, Menezes MP, Mignot C, O'Sullivan J, Orcesi S, Picco PP, Riva E, Robinson RA, Rodriguez D, Salvatici E, Scott C, Szybowska M, Tolmie JL, Vanderver A, Vanhulle C, Vieira JP, Webb K, Whitney RN, Williams SG, Wolfe LA, Zuberi SM, Hur S, Crow YJ. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. 2014;46:503–9. [PMC free article: PMC4004585] [PubMed: 24686847]
- Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U, Barnerias C, Bernard G, Bodemer C, Botella MP, Cereda C, Chandler KE, Dabydeen L, Dale RC, De Laet C, De Goede CG, Del Toro M, Effat L, Enamorado NN, Fazzi E, Gener B, Haldre M, Lin JP, Livingston JH, Lourenco CM, Marques W Jr, Oades P, Peterson P, Rasmussen M, Roubertie A, Schmidt JL, Shalev SA, Simon R, Spiegel R, Swoboda KJ, Temtamy SA, Vassallo G, Vilain CN, Vogt J, Wermenbol V, Whitehouse WP, Soler D, Olivieri I, Orcesi S, Aglan MS, Zaki MS, Abdel-Salam GM, Vanderver A, Kisand K, Rozenberg F, Lebon P, Crow YJ. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013a;12:1159–69. [PMC free article: PMC4349523] [PubMed: 24183309]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin JP, Lourenco CM, Male AM, Marques W Jr, Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O'Connell MA, Lovell SC, Crow YJ. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–8. [PMC free article: PMC4154508] [PubMed: 23001123]
- Rice GI, Reijns MA, Coffin SR, Forte GM, Anderson BH, Szynkiewicz M, Gornall H, Gent D, Leitch A, Botella MP, Fazzi E, Gener B, Lagae L, Olivieri I, Orcesi S, Swoboda KJ, Perrino FW, Jackson AP, Crow YJ. Synonymous mutations in RNASEH2A create cryptic splice sites impairing RNase H2 enzyme function in Aicardi-Goutières syndrome. Hum Mutat. 2013b;34:1066–70. [PMC free article: PMC3714325] [PubMed: 23592335]
- Rice GI, Rodero MP, Crow YJ. Human disease phenotypes associated with mutations in TREX1. J Clin Immunol. 2015;35:235–43. [PubMed: 25731743]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, Spitzer D, Liszewski MK, Barilla-Labarca ML, Terwindt GM, Kasai Y, McLellan M, Grand MG, Vanmolkot KR, de Vries B, Wan J, Kane MJ, Mamsa H, Schafer R, Stam AH, Haan J, de Jong PT, Storimans CW, van Schooneveld MJ, Oosterhuis JA, Gschwendter A, Dichgans M, Kotschet KE, Hodgkinson S, Hardy TA, Delatycki MB, Hajj-Ali RA, Kothari PH, Nelson SF, Frants RR, Baloh RW, Ferrari MD, Atkinson JP. C-terminal truncations in human 3'-5' DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet. 2007;39:1068–70. [PubMed: 17660820]
- Samson JF, Barth PG, de Vries JI, Menko FH, Ruitenbeek W, van Oost BA, Jakobs C. Familial mitochondrial encephalopathy with fetal ultrasonographic ventriculomegaly and intracerebral calcifications. Eur J Pediatr. 1994;153:510–6. [PubMed: 7957369]
- Sanchis A, Cervero L, Bataller A, Tortajada JL, Huguet J, Crow YJ, Ali M, Higuet LJ, Martinez-Frias ML. Genetic syndromes mimic congenital infections. J Pediatr. 2005;146:701–5. [PubMed: 15870678]
- Stephenson JB (2002) Aicardi-Goutieres syndrome - observations of the Glasgow school. Eur J Paediatr Neurol 6 Suppl A:67-70. [PubMed: 12365363]
- Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–98. [PMC free article: PMC2626626] [PubMed: 18724932]
- Thiele H, du Moulin M, Barczyk K, George C, Schwindt W, Nürnberg G, Frosch M, Kurlemann G, Roth J, Nürnberg P, Rutsch F. Cerebral arterial stenoses and stroke: novel features of Aicardi-Goutières syndrome caused by the Arg164X mutation in SAMHD1 are associated with altered cytokine expression. Hum Mutat. 2010;31:E1836–50. [PMC free article: PMC3049152] [PubMed: 20842748]
- Tojo K, Sekijima Y, Suzuki T, Suzuki N, Tomita Y, Yoshida K, Hashimoto T, Ikeda S. Dystonia, mental deterioration, and dyschromatosis symmetrica hereditaria in a family with ADAR1 mutation. Mov Disord. 2006;21:1510–3. [PubMed: 16817193]
- Tolmie JL, Shillito P, Hughes-Benzie R, Stephenson JB. The Aicardi-Goutieres syndrome (familial, early onset encephalopathy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis). J Med Genet. 1995;32:881–4. [PMC free article: PMC1051740] [PubMed: 8592332]
- Uggetti C, La Piana R, Orcesi S, Egitto MG, Crow YJ, Fazzi E. Aicardi-Goutieres syndrome: neuroradiologic findings and follow-up. AJNR Am J Neuroradiol. 2009;30:1971–6. [PMC free article: PMC7051307] [PubMed: 19628626]
- Xin B, Jones S, Puffenberger EG, Hinze C, Bright A, Tan H, Zhou A, Wu G, Vargus-Adams J, Agamanolis D, Wang H. Homozygous mutation in SAMHD1 gene causes cerebral vasculopathy and early onset stroke. Proc Natl Acad Sci U S A. 2011;108:5372–7. [PMC free article: PMC3069167] [PubMed: 21402907]
- Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. 2007;131:873–86. [PubMed: 18045533]
Publication Details
Author Information and Affiliations
Institut Imagine
Paris, France
University of Manchester
Manchester, United Kingdom
Publication History
Initial Posting: June 29, 2005; Last Update: November 22, 2016.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Crow YJ. Aicardi-Goutières Syndrome. 2005 Jun 29 [Updated 2016 Nov 22]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.


