Summary
Clinical characteristics.
The creatine deficiency disorders (CDDs), inborn errors of creatine metabolism and transport, comprise three disorders: the creatine biosynthesis disorders guanidinoacetate methyltransferase (GAMT) deficiency and L-arginine:glycine amidinotransferase (AGAT) deficiency; and creatine transporter (CRTR) deficiency. Developmental delay and cognitive dysfunction or intellectual disability and speech-language disorder are common to all three CDDs.
- Onset of clinical manifestations of GAMT deficiency (reported in ~130 individuals) is between ages three months and two years; in addition to developmental delays, the majority of individuals have epilepsy and develop a behavior disorder (e.g., hyperactivity, autism, or self-injurious behavior), and about 30% have movement disorder.
- AGAT deficiency has been reported in 16 individuals; none have had epilepsy or movement disorders.
- Clinical findings of CRTR deficiency in affected males (reported in ~130 individuals) in addition to developmental delays include epilepsy (variable seizure types and may be intractable) and behavior disorders (e.g., attention deficit and/or hyperactivity, autistic features, impulsivity, social anxiety), hypotonia, and (less commonly) a movement disorder. Poor weight gain with constipation and prolonged QTc on EKG have been reported. While mild-to-moderate intellectual disability is commonly observed up to age four years, the majority of adult males with CRTR deficiency have been reported to have severe intellectual disability. Females heterozygous for CRTR deficiency are typically either asymptomatic or have mild intellectual disability, although a more severe phenotype resembling the male phenotype has been reported.
Diagnosis/testing.
The diagnosis of a CDD is established in a proband with suggestive findings and biallelic pathogenic variants in GAMT or GATM or a hemizygous or heterozygous pathogenic variant in SLC6A8 identified by molecular genetic testing.
Management.
Treatment of manifestations: GAMT deficiency and AGAT deficiency are treated with oral creatine monohydrate to replenish cerebral creatine levels. Treatment of GAMT deficiency requires supplementation of ornithine and dietary restriction of arginine or protein. CRTR deficiency is treated with oral creatine monohydrate and arginine and glycine supplementation. The developmental delay, intellectual disability, and behavior problems are managed with an individualized education and therapy program; epilepsy and movement disorder are treated by the appropriate specialist in a standard manner.
Surveillance: In those treated with creatine monohydrate, periodic determination of cerebral creatine level by in vivo 1H-MRS and annual measurement of renal function to detect possible creatine-associated nephropathy is warranted. Developmental and neurologic assessments are recommended at each clinic visit.
Evaluation of relatives at risk: Early diagnosis of neonates at risk for a CDD by biochemical or molecular genetic testing allows for early diagnosis and treatment.
Genetic counseling.
GAMT deficiency (caused by pathogenic variants in GAMT) and AGAT deficiency (caused by pathogenic variants in GATM) are inherited in an autosomal recessive manner. CRTR deficiency (caused by pathogenic variants in SLC6A8) is inherited in an X-linked manner.
- Autosomal recessive inheritance. If both parents are known to be heterozygous for a GAMT or GATM pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the GAMT or GATM pathogenic variants have been identified in an affected family member, molecular genetic carrier testing and prenatal and preimplantation genetic testing are possible.
- X-linked inheritance. Mothers who are heterozygous for an SLC6A8 pathogenic variant have a 50% chance of transmitting the pathogenic variant in each pregnancy; sons who inherit the pathogenic variant will be affected; daughters who inherit the pathogenic variant will be heterozygotes and may develop clinical findings related to the disorder. Once the SLC6A8 pathogenic variant has been identified in an affected family member, molecular genetic testing to identify female heterozygotes and prenatal and preimplantation genetic testing are possible.
GeneReview Scope
Table
Guanidinoacetate methyltransferase (GAMT) deficiency L-arginine:glycine amidinotransferase (AGAT) deficiency
Diagnosis
The creatine deficiency disorders (CDDs) are inborn errors of creatine metabolism and transport that comprise:
- Two creatine biosynthesis defects (both inherited in an autosomal recessive manner):
- Guanidinoacetate methyltransferase (GAMT) deficiency
- L-arginine:glycine amidinotransferase (AGAT) deficiency
- One creatine transporter defect (inherited in an X-linked manner): creatine transporter (CRTR) deficiency
Suggestive Findings
A CDD should be suspected in probands with the following clinical, biochemical, and imaging findings and family history.
Clinical findings
- Developmental delay
- Cognitive dysfunction or intellectual disability
- Hypotonia
- Seizures or refractory epilepsy
- Movement disorders (e.g., chorea-athetosis, dystonia)
- Behavior problems (e.g., attention-deficit/hyperactivity disorder, autism spectrum disorder, aggressive behavior)
Biochemical findings [van de Kamp et al 2014, Mørkrid et al 2015]
- GAMT deficiency. Elevated guanidinoacetate (GAA) levels in urine, plasma, or cerebrospinal fluid (CSF) and low or low-normal creatine and creatinine levels in urine, plasma, or CSF
- AGAT deficiency. Low GAA levels in urine, plasma, or CSF and low or low-normal creatine and creatinine levels in urine, plasma, or CSF
- CRTR deficiency. Elevated creatine-to-creatinine ratio in urine in males. Females can have normal or mildly elevated creatine-to-creatinine ratio in urine.
Imaging findings
- Proton magnetic resonance spectroscopy (1H-MRS) reveals absent or significantly decreased creatine peak in the brain in all individuals with GAMT deficiency and AGAT deficiency and in males with CRTR deficiency [van de Kamp et al 2014].
- 1H-MRS reveals partial depletion or normal levels of creatine peak in the brain in heterozygous females with X-linked CRTR deficiency [van de Kamp et al 2011].
Family history
- GAMT and AGAT deficiencies. Consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.
- CRTR deficiency. Consistent with X-linked inheritance (e.g., no male-to-male transmission, affected maternal uncle or male sibs, normal to mildly affected female sibs or mother). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of a CDD is established in a proband with suggestive clinical findings by identification of biallelic pathogenic (or likely pathogenic) variants in GAMT or GATM or of a hemizygous or heterozygous pathogenic (or likely pathogenic) variant in SLC6A8 on molecular genetic testing.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic GAMT or GATM variants of uncertain significance (or of one known pathogenic variant and one variant of uncertain significance) does not establish or rule out the diagnosis of GAMT or AGAT deficiency. In these individuals, typical brain 1H-MRS and abnormal urine, plasma, or CSF GAA and creatine levels and GAMT and AGAT activity measurements in fibroblasts or in lymphocytes will be required to confirm biochemical diagnosis of GAMT or AGAT deficiency. (3) Identification of a hemizygous or heterozygous SLC6A8 variant of uncertain significance does not establish or rule out a diagnosis of CTR deficiency. In these individuals, abnormal brain 1H-MRS and abnormal levels of urine creatine-to-creatinine ratio in males will be required to confirm biochemical diagnosis. Creatine uptake in the individual's cultured skin fibroblasts will be required to confirm biochemical diagnosis of CRTR deficiency in males.
A diagnostic testing algorithm is useful for guiding molecular genetic testing in an individual suspected of having suggestive clinical, biochemical, and/or imaging findings and/or reduced creatine levels on brain 1H-MRS (see Figure 1).
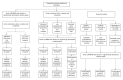
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel for epilepsy, intellectual disability, or autism spectrum disorder) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved based on the biochemical and imaging findings, whereas genomic testing does not. Individuals with the distinctive biochemical or imaging findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other inherited disorders with intellectual disability and/or epilepsy are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of the gene predicted from the diagnostic testing algorithm is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If only one (for GAMT or GATM) or no variant (for any of the 3 genes) is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
A multigene panel for epilepsy, intellectual disability, or autism spectrum disorder that includes GAMT, GATM, SLC6A8, and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Creatine Deficiency Disorder
Clinical Characteristics
Clinical Description
Developmental delay, cognitive dysfunction, and intellectual disability are common to all three creatine deficiency disorders (CDDs). See Table 2 for comparison of the three deficiencies; further details follow the table.
Table 2.
Creatine Deficiency Disorders: Comparison of Phenotypes by Select Features
GAMT Deficiency
To date, about 130 individuals have been identified with biallelic pathogenic variants in GAMT [Stockler-Ipsiroglu et al 2014, Khaikin et al 2018]. The following description of the phenotypic features associated with this condition is based on these reports.
Onset of the first clinical manifestations ranges from early infancy (age 3-6 months) to age two years [Khaikin et al 2018]. The age of diagnosis ranges from neonatal to 34 years [Stockler-Ipsiroglu et al 2014, Khaikin et al 2018].
Developmental delay (DD) and cognitive dysfunction or intellectual disability (ID), the most consistent clinical manifestation, is present in all affected individuals. The severity ranges from mild to severe. About 50%-75% of individuals with GAMT deficiency have severe DD or ID [Mercimek-Mahmutoglu et al 2014, Stockler-Ipsiroglu et al 2014, Khaikin et al 2018].
Speech-language disorder. Variable expressive language defects were reported in two sibs with GAMT deficiency: the proband spoke fewer than ten words, whereas her younger sister spoke in short sentences at age 13 years [O'Rourke et al 2009].
A behavior disorder (e.g., hyperactivity, autism, or self-injurious behavior) is reported in more than 75% of affected individuals [Mercimek-Mahmutoglu et al 2014, Khaikin et al 2018].
Seizures, the third most consistent manifestation in GAMT deficiency, are observed in more than 70% of affected individuals. Seizure types include myoclonic, generalized tonic-clonic, partial complex, head nodding, and atonic seizures. Seizure severity ranges from occasional seizures to seizures that are non-responsive to various anti-seizure medications [Mercimek-Mahmutoglu et al 2014, Stockler-Ipsiroglu et al 2014, Khaikin et al 2018].
Movement disorders, observed in about 30% of individuals, are mainly chorea, athetosis, dystonia, or ataxia [Mercimek-Mahmutoglu et al 2014, Stockler-Ipsiroglu et al 2014, Khaikin et al 2018]. Pathologic signal intensities in the basal ganglia in brain MRI are observed in individuals with or without a movement disorder [Mercimek-Mahmutoglu et al 2014, Stockler-Ipsiroglu et al 2014, Khaikin et al 2018]. The onset is usually before age 12 years; however, a young woman with GAMT deficiency was reported to have onset of a movement disorder (including ballistic and dystonic movements) at age 17 years [O'Rourke et al 2009].
AGAT Deficiency
To date, 16 individuals have been identified with biallelic pathogenic variants in GATM [Stockler-Ipsiroglu et al 2015]. The following description of the phenotypic features associated with this condition is based on these reports.
DD and cognitive dysfunction or ID, the most consistent clinical manifestation, is present in all affected individuals. The severity of intellectual disability ranges from mild to severe, although more than 80% of the individuals have mild-to-moderate intellectual disability.
A single seizure, observed in about 10% of affected individuals, was reported to occur with or without fever.
Movement disorders were not reported in any affected individuals.
A behavior disorder was present in 25% of affected individuals.
Muscle weakness / myopathy was observed in 50% of affected individuals.
CRTR Deficiency ‒ Affected Males
To date, about 130 individuals have been identified with a pathogenic variant in SLC6A8 [van de Kamp et al 2013a, Bruun et al 2018, Bahl et al 2020]. The following description of the phenotypic features associated with this condition is based on these reports.
Onset of the first clinical manifestations ranges from four to 54 months [Bruun et al 2018]. The age at diagnosis ranges from one to 66 years, indicating that life expectancy can be normal. Now that the disorder is reasonably well described and diagnostic testing is more widely available, it is anticipated that diagnosis will mainly occur within the first three years of life.
DD and cognitive dysfunction or ID was present in all affected male individuals ranging from mild to severe: 85% of affected males had mild-to-moderate ID up to age four years; 75% of affected males older than age 18 years had severe ID [van de Kamp et al 2013a]. One adult had progressive cognitive dysfunction [Kleefstra et al 2005].
Speech-language disorder. Speech development was delayed in all affected males. First words were at a mean age of 3.1 years (age range: 9 months to 10 years). In affected males older than age ten years, 14% had no speech development, 55% were able to speak single words, and 31% were able to speak in sentences [van de Kamp et al 2013a].
A neuropsychological profile in four affected boys from two unrelated families from the Netherlands revealed a semantic-pragmatic language disorder (difficulty in understanding the meaning of words) with oral dyspraxia [Mancini et al 2005].
Seizures were present in 59% of affected male individuals. The most common seizure types were generalized tonic-clonic and simple or complex partial seizures with or without secondary generalization. Absence and myoclonic seizures were rare. Seizure onset was between ages one and 21 years [van de Kamp et al 2013b]. Intractable epilepsy has been reported in fewer than ten individuals [Mercimek-Mahmutoglu et al 2010, van de Kamp et al 2013a].
Movement disorder. Wide-based gait or ataxia and dystonia or athetosis were reported in 29% and 11% of affected males, respectively [van de Kamp et al 2013a].
Behavior disorder. Behavior disorder was reported in 85% of affected males. The most common behavior disorders were attention deficit and/or hyperactivity (55%) and autistic features (41%). Other behavior disorders reported in affected males include social anxiety or shyness (20%), stereotypic behavior (20%), impulsive behavior (27%), aggressive behavior (19%), self-injurious behavior (10%), and obsessive-compulsive behavior (8%) [van de Kamp et al 2013a].
Other neurologic clinical features. Hypotonia was present in 40% of affected males. Spasticity was reported in 26% of affected males. Four individuals had mild (sensorineural) hearing loss. Nine affected males were reported with strabismus or bilateral abducens nerve palsy. Myopathic face, ptosis, joint laxity (likely secondary to the hypotonia), and decreased muscle bulk were also reported [van de Kamp et al 2013a].
Other non-neurologic clinical features
- Dysmorphic features including microcephaly, broad forehead, midface retrusion, high palate, short nose, prominent nasal bridge, ear differences (underfolded helices, large ears, and/or cupped ears), deeply set eyes, fifth finger clinodactyly, and slender body build were reported in 45% of affected males [van de Kamp et al 2013a, van de Kamp et al 2013b].
- Gastrointestinal findings, including poor weight gain, vomiting, constipation, ileus (likely secondary to constipation), hepatitis, gastric and duodenal ulcers, and hiatal hernia (which may or may not be related to CRTR deficiency) were reported in 35% of affected males [van de Kamp et al 2013a].
- Cardiac features. One affected male had long QT disorder [van de Kamp et al 2013a]. Seven males with CRTR deficiency (39%) had prolonged QTc on EKG. These individuals also showed increased left ventricular internal dimension (diastole) and diminished left ventricular posterior wall dimension (diastole) in echocardiography [Levin et al 2021].
- Medical concerns in adulthood. Twenty-one of 101 affected males were adults (age >18 years). They presented with myopathic face, ptosis, external ophthalmoplegia, or parkinsonism. Chronic constipation leading to megacolon, ileus, or bowel perforation and/or gastric or duodenal ulcer disease have been reported in some adults [van de Kamp et al 2013a].
CRTR Deficiency ‒ Heterozygous Females
Females heterozygous for their family-specific SLC6A8 pathogenic variant are typically either asymptomatic or have mild ID [van de Kamp et al 2011]. There was no clinical correlation between skewed X-chromosome inactivation in favor of the pathogenic variant allele and severity of clinical phenotype. There was no significant statistical correlation between intellectual ability and cerebral creatine level on brain 1H-MRS [van de Kamp et al 2011]. A female with mild ID, intractable epilepsy, and behavior problems (a phenotype similar to affected males) did not have evidence of skewed X-chromosome inactivation in peripheral blood cells; tissue-specific skewed X-chromosome inactivation in the brain could explain her severe neurologic findings [Mercimek-Mahmutoglu et al 2010].
Genotype-Phenotype Correlations
No genotype-phenotype correlations for any of the CDDs have been identified.
Prevalence
GAMT deficiency. About 130 individuals with GAMT deficiency have been diagnosed worldwide.
The estimated incidence of GAMT deficiency in the general population ranges from 1:2,640,000 to 1:250,000 [Desroches et al 2015, Mercimek-Mahmutoglu et al 2016]. This is in agreement with information from pilot newborn screening programs for GAMT deficiency, which screened more than 1,500,000 newborns; to date two of the newborns have a confirmed diagnosis of GAMT deficiency [Mercimek-Mahmutoglu et al 2012, Pasquali et al 2014, Pitt et al 2014, Stockler-Ipsiroglu et al 2014, Sinclair et al 2016, Hart et al 2021].
The estimated incidence of GAMT deficiency in the Utah and New York newborn population was 1:405,655 [Hart et al 2021].
Smaller studies of individuals with neurologic disease or severe intellectual disability found GAMT deficiency present in 1.1% [Cheillan et al 2012].
AGAT deficiency. The estimated carrier frequency of AGAT deficiency was one in 1,292 (0.077%; CI=0.06%-0.10%) in the general population using the ExAC Browser Beta database [DesRoches et al 2016].
CRTR deficiency. CRTR deficiency has been studied in many cohorts ranging from 49 to 4,426 individuals with familial or nonfamilial ID. These studies were summarized by van de Kamp et al [2014]; the prevalence in males with ID was estimated between 0.4% and 1.4%. In a recent study, the prevalence of creatine transporter deficiency was 2.64% in individuals with neurodevelopmental disorders [Bahl et al 2020].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in GAMT, GATM, or SLC6A8.
Differential Diagnosis
Disorders summarized in Table 3 should be considered in individuals with partial creatine deficiency in the brain detected by 1H-MRS, who have normal concentrations of guanidinoacetate (GAA) in the urine, plasma, and CSF and a normal creatine-to-creatinine ratio in urine.
Table 3.
Disorders of Interest in the Differential Diagnosis of Creatine Deficiency Disorders
Management
No clinical practice guidelines for creatine deficiency disorders (CDDs) have been published. Some of the references list management recommendations for GAMT and CRTR deficiencies [Stockler-Ipsiroglu et al 2014, Bruun et al 2018, Khaikin et al 2018].
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with a CDD, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with Creatine Deficiency Disorders
Treatment of Manifestations
The general treatment of CDDs of any cause is presented in Table 5.
Medical treatment specific to the type of CDD follows in Table 6 (GAMT deficiency), Table 7 (AGAT deficiency), and Table 8 (CRTR deficiency).
Table 5.
Treatment of Manifestations in Individuals with Creatine Deficiency Disorders
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
- For muscle tone abnormalities including hypertonia or dystonia, consider involving appropriate specialists to aid in management of baclofen, tizanidine, Botox®, anti-parkinsonian medications, or orthopedic procedures.
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Social/Behavioral Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder, when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Table 6.
Treatment of Manifestations in Individuals with GAMT Deficiency
Table 7.
Treatment of Manifestations in Individuals with AGAT Deficiency
Table 8.
Treatment of Manifestations in Individuals with CRTR Deficiency
Prevention of Primary Manifestations
Surveillance
Surveillance recommendations for CDDs are summarized in Table 9.
Table 9.
Recommended Surveillance for Individuals with Creatine Deficiency Disorders
Evaluation of Relatives at Risk
It is appropriate to evaluate neonates at risk for a creatine deficiency disorder to allow for early diagnosis and treatment. (Note: It is not proven that early treatment of CRTR deficiency would change the outcome.)
Evaluations can include:
- Molecular genetic testing if the pathogenic variants in the family are known;
- Biochemical genetic testing if the pathogenic variants in the family are not known.
See Genetic Counseling for issues related to evaluation of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Pregnancy management has been reported in a single individual with AGAT deficiency. The fetal growth and head circumference declined by 20 weeks of pregnancy during monitoring. Creatine levels were low in the pregnant woman. Daily creatine dose was increased in the pregnant woman, and the baby was normal at delivery and was not affected with AGAT deficiency [Alessandrì et al 2020].
Therapies Under Investigation
There are no current clinical trials for any of the CDDs. Some pharmacotherapies are being investigated in cell lines or animal models of CRTR deficiency:
- Swiss mice brain hippocampal slices were treated with diacetyl creatine ethyl ester (DAC) after creatine transporter was blocked using guanidinopropionic acid. The study showed an increase in intracellular creatine, and DAC was metabolized to creatine. Authors concluded that further research is needed to fully elucidate their hypotheses [Adriano et al 2018].
- Dodecyl creatine ester was administered intranasally and intracerebroventricularly in Slc6a8-/y mice and a novel object recognition (NOR) test was applied. There was restoration of NOR learning and 50% increase in creatine [Ullio-Gamboa et al 2019].
- 4-phenylbutyrate (4-PBA) was shown to increase creatine uptake in transfected HEK293 cells expressing pathogenic variants in SLC6A8 [El-Kasaby et al 2019]. 4-PBA was considered to be a pharmaco-chaperone to rescue CTRT activity.
- Cyclocreatine was used in a CRTR deficiency mouse model and was effective in treating cognitive functions, epilepsy, and behavioral features [Cacciante et al 2020].
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Guanidinoacetate methyltransferase (GAMT) deficiency and L-arginine:glycine amidinotransferase (AGAT) deficiency are inherited in an autosomal recessive manner.
Creatine transporter (CRTR) deficiency is inherited in an X-linked manner.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
- The parents of a child with GAMT or AGAT deficiency are presumed to be heterozygous for a GAMT or GATM pathogenic variant.
- Molecular genetic testing recommended for the parents of a proband to confirm that both parents are heterozygous for a GAMT or GATM pathogenic variant and to allow reliable recurrence risk assessment.
- If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, it is possible that one of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017]. If the proband appears to have homozygous pathogenic variants (i.e., the same 2 pathogenic variants), additional possibilities to consider include:
- A single- or multiexon deletion in the proband that was not detected by sequence analysis and that resulted in the artifactual appearance of homozygosity;
- Uniparental isodisomy for the parental chromosome with the pathogenic variant that resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for a GAMT or GATM pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. To date, individuals with GAMT or AGAT deficiency are not known to reproduce.
Other family members. Each sib of the proband's parents is at 50% risk of being a carrier of a GAMT or GATM pathogenic variant.
Carrier detection. Molecular genetic carrier testing for at-risk relatives requires prior identification of the GAMT or GATM pathogenic variants in the family.
Note: Because biochemical testing is normal in carriers for GAMT and AGAT deficiencies, identification of carriers requires molecular genetic testing for the familial GAMT or GATM pathogenic variants.
X-Linked Inheritance – Risk to Family Members
Parents of a male proband
- The father of a male with CRTR deficiency will not have the disorder nor will he be hemizygous for the SLC6A8 pathogenic variant; therefore, he does not require further evaluation/testing.
- In a family with more than one affected child, the mother of an affected male is an obligate heterozygote. Note: If a woman has more than one affected child and no other affected relatives and if the SLC6A8 pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism.
- If a male is the only affected family member, the mother may be a heterozygote, the affected male may have a de novo SLC6A8 pathogenic variant (in which case the mother is not a heterozygote), or the mother may have somatic/germline mosaicism [van de Kamp et al 2013a, van de Kamp et al 2014].
- SLC6A8 pathogenic variants occurred de novo in 30% of the individuals with CRTR deficiency in a retrospective study of 85 families [van de Kamp et al 2013a].
- 7% of mothers in a retrospective study of 85 families had somatic/germline mosaicism [van de Kamp et al 2013a].
- Molecular genetic testing of the mother is recommended to confirm her genetic status and to allow reliable recurrence risk assessment.
- Heterozygous mothers may have a history of learning disability or mild intellectual disability or seizures [Mercimek-Mahmutoglu et al 2010, van de Kamp et al 2011] (see Clinical Description, CRTR Deficiency ‒ Heterozygous Females).
Sibs of a male proband. The risk to sibs depends on the genetic status of the mother:
- If the mother of the proband has an SLC6A8 pathogenic variant, the chance of transmitting it in each pregnancy is 50%:
- Males who inherit the pathogenic variant will be affected;
- Females who inherit the pathogenic variant will be heterozygous and may develop clinical findings related to the disorder (see Clinical Description, CRTR Deficiency ‒ Heterozygous Females).
- If the proband represents a simplex case (i.e., a single occurrence in a family) and if the SLC6A8 pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is presumed to be low but greater than that of the general population because of the possibility of maternal germline mosaicism [van de Kamp et al 2011, van de Kamp et al 2013a].
Offspring of a male proband. Affected males are not to known to reproduce.
Other family members. The proband's maternal aunts may be at risk of being heterozygous for the SLC6A8 pathogenic variant and the aunts' offspring, depending on their sex, may be at risk of being heterozygous or hemizygous for the pathogenic variant.
Heterozygote detection. Molecular genetic testing to identify female heterozygotes requires prior identification of the SLC6A8 pathogenic variant in the family. Note: Females who are heterozygous for an SLC6A8 pathogenic variant may develop clinical findings related to the disorder (see Clinical Description, CRTR Deficiency ‒ Heterozygous Females).
Note: Because heterozygous females may have a normal creatine-to-creatinine ratio in urine and normal creatine content on brain 1H-MRS [van de Kamp et al 2011], identification of female heterozygotes requires SLC6A8 molecular genetic testing.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are heterozygous or are at risk of being heterozygous.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the GAMT, GATM, or SLC6A8 pathogenic variant(s) have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Biochemical testing. Prenatal diagnosis for a pregnancy at increased risk for GAMT deficiency is possible by analysis of guanidinoacetate (GAA) and creatine in amniotic fluid. Amniocentesis for prenatal diagnosis was performed at 15 weeks' gestation in the mother of a child with GAMT deficiency. GAA was 11.43 μmol/L (normal range for 15 weeks of amenorrhea was 2.96±0.70 μmol/L) [Cheillan et al 2006]. As there is very limited information on the use of biochemical testing in the prenatal diagnosis of creatine deficiency disorders, molecular genetic testing is the preferred method for prenatal diagnosis.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Association for Creatine Deficiencies1024 Bayside DriveSuite 532Newport Beach CA 92660Email: kim@creatineinfo.org
- MedlinePlus
- MedlinePlus
- MedlinePlus
- American Association on Intellectual and Developmental Disabilities (AAIDD)Phone: 202-387-1968
- American Epilepsy Society
- Epilepsy FoundationPhone: 800-332-1000; 866-748-8008
- Metabolic Support UKUnited KingdomPhone: 0845 241 2173
- Association for Creatine Deficiencies Patient Registry
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Creatine Deficiency Disorders: Genes and Databases
Table B.
OMIM Entries for Creatine Deficiency Disorders (View All in OMIM)
Molecular Pathogenesis
Creatine is synthesized by two enzymatic reactions:
- Transfer of the amidino group from arginine to glycine, yielding guanidinoacetic acid (GAA) and catalyzed by L-arginine:glycine amidinotransferase (also known as glycine amidinotransferase, mitochondrial, AGAT, or GATM)
- Methylation of the amidino group in the GAA molecule by S-adenosyl-L-methionine:N-guanidinoacetate methyltransferase (also known as guanidinoacetate N-methyltransferase or GAMT)
Creatine is synthesized primarily in the kidney and pancreas, which have high AGAT activity, and in the liver, which has high GAMT activity. Both genes and enzymes have been detected in the brain as well [Braissant & Henry 2008].
Synthesized creatine is transported via the bloodstream to the organs of utilization (mainly muscle and brain), where it is taken up via sodium- and chloride-dependent creatine transporter 1 (SLC6A8 protein) (Figure 2) [Wyss & Kaddurah-Daouk 2000]. This protein is predominantly expressed in skeletal muscle and kidney, but also found in brain, heart, colon, testis, and prostate. The creatine-phosphocreatine shuttle has a key function in the maintenance of the energy supply to skeletal and cardiac muscle. Muscle cells do not synthesize creatine, but take it up via a special sodium-dependent transporter, the creatine transporter.
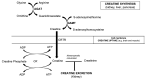
Mechanism of disease causation
- Biallelic pathogenic variants in either GAMT or GATM result in a deficiency of the associated enzyme, resulting in deficient synthesis of creatine.
- A hemizygous (or heterozygous) pathogenic variant in SLC6A8 results in deficient synthesis of the creatine transporter protein, resulting in deficient transport of creatine to the brain and muscle.
Gene-specific laboratory considerations: SLCA8. A paralogous copy of this gene is localized at chromosome 16, which can interfere with the genetic tests.
Chapter Notes
Author Notes
Dr Mercimek-Andrews has long-standing experience in the diagnosis and treatment of creatine deficiency disorders (CDDs). She is a neurometabolic physician and clinician scientist. She treats patients with CRTR deficiency and provides consultations to physicians and parents for the treatment of CDDs. She is one of the Scientific Medical Advisory Board members for the Association for Creatine Deficiencies.
www.wchri.org/members-and-trainees/find-a-researcher/saadet-andrews/
www.ualberta.ca/medical-genetics/people/faculty/saadet-andrews.html
Dr Salomons has long-standing experience in the laboratory diagnosis of CDDs. She is a clinical laboratory genetic specialist and head of the Laboratory of Genetic Metabolic Diseases at the Amsterdam University Medical Centers, the Netherlands. She discovered CRTR deficiency with her team and that of Dr DeGrauw.
www.amc.nl/web/laboratory-genetic-metabolic-diseases-lgmd.htm
Acknowledgments
We would like to thank Dr Sylvia Stöckler (Stöckler-Ipsiroglu), who discovered the first two creatine biosynthesis defects, GAMT deficiency and AGAT deficiency.
We would like to thank Dr DeGrauw and Dr Salomons, who discovered CRTR deficiency.
We would like to thank Dr Schulze, who discovered the high-dose ornithine supplementation combined with an arginine- and protein-restricted diet for the treatment of GAMT deficiency.
Author History
Saadet Mercimek-Andrews, MD, PhD, FCCMG, FRCPC (2008-present)
Gaija S Salomons, PhD (2008-present)
Sylvia Stöckler-Ipsiroglu, MD, PhD, MBA, FRCPC; University of British Columbia (2008-2015)
Revision History
- 10 February 2022 (ha) Comprehensive update posted live
- 10 December 2015 (me) Comprehensive update posted live
- 18 August 2011 (me) Comprehensive update posted live
- 15 January 2009 (me) Review posted live
- 24 July 2008 (smm) Original submission
References
Literature Cited
- Adriano E, Gulino M, Arkel M, Salis A, Damonte G, Liessi N, Millo E, Garbati P, Balestrino M. Di-acetyl creatine ethyl ester, a new creatine derivative for the possible treatment of creatine transporter deficiency. Neurosci Lett. 2018;665:217–23. [PubMed: 29229397]
- Alessandrì MG, Strigini F, Cioni G, Battini R. Increased creatine demand during pregnancy in Arginine: Glycine Amidino-Transferase deficiency: a case report. BMC Pregnancy Childbirth. 2020;20:506. [PMC free article: PMC7469264] [PubMed: 32883247]
- Bahl S, Cordeiro D, MacNeil L, Schulze A, Mercimek-Andrews S. Urine creatine metabolite panel as a screening test in neurodevelopmental disorders. Orphanet J Rare Dis. 2020;15:339. [PMC free article: PMC7709238] [PubMed: 33267903]
- Battini R, Alessandrì MG, Leuzzi V, Moro F, Tosetti M, Bianchi MC, Cioni G. Arginine:glycine amidinotransferase (AGAT) deficiency in a newborn: early treatment can prevent phenotypic expression of the disease. J Pediatr. 2006;148:828–30. [PubMed: 16769397]
- Battini R, Alessandrì MG, Casalini C, Casarano M, Tosetti M, Cioni G. Fifteen-year follow-up of Italian families affected by arginine glycine amidinotransferase deficiency. Orphanet J Rare Dis. 2017;12:21. [PMC free article: PMC5289057] [PubMed: 28148286]
- Boenzi S, Pastore A, Martinelli D, Goffredo BM, Boiani A, Rizzo C, Dionisi-Vici C. Creatine metabolism in urea cycle defects. J Inherit Metab Dis. 2012;35:647–53. [PubMed: 22644604]
- Braissant O, Henry H. AGAT, GAMT and SLC6A8 distribution in the central nervous system, in relation to creatine deficiency syndromes: a review. J Inherit Metab Dis. 2008;31:230–9. [PubMed: 18392746]
- Bruun TUJ, Sidky S, Bandeira AO, Debray FG, Ficicioglu C, Goldstein J, Joost K, Koeberl DD, Luísa D, Nassogne MC, O'Sullivan S, Õunap K, Schulze A, van Maldergem L, Salomons GS, Mercimek-Andrews S. Treatment outcome of creatine transporter deficiency: international retrospective cohort study. Metab Brain Dis. 2018;33:875–84. [PubMed: 29435807]
- Cacciante F, Gennaro M, Sagona G, Mazziotti R, Lupori L, Cerri E, Putignano E, Butt M, Do MT, McKew JC, Alessandrì MG, Battini R, Cioni G, Pizzorusso T, Baroncelli L. Cyclocreatine treatment ameliorates the cognitive, autistic and epileptic phenotype in a mouse model of creatine transporter deficiency. Sci Rep. 2020;10:18361. [PMC free article: PMC7591530] [PubMed: 33110151]
- Cheillan D, Joncquel-Chevalier Curt M, Briand G, Salomons GS, Mention-Mulliez K, Dobbelaere D, Cuisset JM, Lion-François L, Portes VD, Chabli A, Valayannopoulos V, Benoist JF, Pinard JM, Simard G, Douay O, Deiva K, Afenjar A, Héron D, Rivier F, Chabrol B, Prieur F, Cartault F, Pitelet G, Goldenberg A, Bekri S, Gerard M, Delorme R, Tardieu M, Porchet N, Vianey-Saban C, Vamecq J. Screening for primary creatine deficiencies in French patients with unexplained neurological symptoms. Orphanet J Rare Dis. 2012;7:96. [PMC free article: PMC3552865] [PubMed: 23234264]
- Cheillan D, Salomons GS, Acquaviva C, Boisson C, Roth P, Cordier MP, François L, Jakobs C, Vianey-Saban C. Prenatal diagnosis of guanidinoacetate methyltransferase deficiency: increased guanidinoacetate concentrations in amniotic fluid. Clin Chem. 2006;52:775–7. [PubMed: 16595836]
- Desroches CL, Patel J, Wang P, Minassian B, Marshall CR, Salomons GS, Mercimek-Mahmutoglu S. Carrier frequency of guanidinoacetate methyltransferase deficiency in the general population by functional characterization of missense variants in the GAMT gene. Mol Genet Genomics. 2015;290:2163–71. [PubMed: 26003046]
- DesRoches CL, Bruun T, Wang P, Marshall CR, Mercimek-Mahmutoglu S. Arginine-glycine amidinotransferase deficiency and functional characterization of missense variants in GATM. Hum Mutat. 2016;37:926–32. [PubMed: 27233232]
- El-Kasaby A, Kasture A, Koban F, Hotka M, Asjad HMM, Kubista H, Freissmuth M, Sucic S. Rescue by 4-phenylbutyrate of several misfolded creatine transporter-1 variants linked to the creatine transporter deficiency disorder. Neuropharmacology. 2019;161:107572. [PubMed: 30885608]
- Hart K, Rohrwasser A, Wallis H, Golsan H, Shao J, Anderson T, Wang X, Szabo-Fresnais N, Morrissey M, Kay DM, Wojcik M, Galvin-Parton PA, Longo N, Caggana M, Pasquali M. Prospective identification by neonatal screening of patients with guanidinoacetate methyltransferase deficiency. Mol Genet Metab. 2021;134:60–4. [PubMed: 34389248]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Khaikin Y, Sidky S, Abdenur J, Anastasi A, Ballhausen D, Buoni S, Chan A, Cheillan D, Dorison N, Goldenberg A, Goldstein J, Hofstede FC, Jacquemont ML, Koeberl DD, Lion-Francois L, Lund AM, Mention K, Mundy H, O'Rourke D, Pitelet G, Raspall-Chaure M, Tassini M, Billette de Villemeur T, Williams M, Salomons GS, Mercimek-Andrews S. Treatment outcome of twenty-two patients with guanidinoacetate methyltransferase deficiency: an international retrospective cohort study. Eur J Paediatr Neurol. 2018;22:369–79. [PubMed: 29506905]
- Kleefstra T, Rosenberg EH, Salomons GS, Stroink H, van Bokhoven H, Hamel BC, de Vries BB. Progressive intestinal, neurological and psychiatric problems in two adult males with cerebral creatine deficiency caused by an SLC6A8 mutation. Clin Genet. 2005;68:379–81. [PubMed: 16143026]
- Levin MD, Bianconi S, Smith A, Cawley NX, Do AD, Hammond D, Grafstein JF, Thurm A, Miller J, Perreault J, Noguchi A, Springer D, Kozel BA, Spurney CF, Wassif CA, Yu ZX, Schulze A, Porter FD, Hannah-Shmouni F. X-linked creatine transporter deficiency results in prolonged QTc and increased sudden death risk in humans and disease model. Genet Med. 2021;23:1864–72. [PMC free article: PMC8487919] [PubMed: 34050321]
- Mancini GM, Catsman-Berrevoets CE, de Coo IF, Aarsen FK, Kamphoven JH, Huijmans JG, Duran M, van der Knaap MS, Jakobs C, Salomons GS. Two novel mutations in SLC6A8 cause creatine transporter defect and distinctive X-linked mental retardation in two unrelated Dutch families. Am J Med Genet A. 2005;132A:288–95. [PubMed: 15690373]
- Martinelli D, Häberle J, Rubio V, Giunta C, Hausser I, Carrozzo R, Gougeard N, Marco-Marín C, Goffredo BM, Meschini MC, Bevivino E, Boenzi S, Colafati GS, Brancati F, Baumgartner MR, Dionisi-Vici C. Understanding pyrroline-5-carboxylate synthetase deficiency: clinical, molecular, functional, and expression studies, structure-based analysis, and novel therapy with arginine. J Inherit Metab Dis. 2012;35:761–6. [PubMed: 22170564]
- Mercimek-Mahmutoglu S, Connolly MB, Poskitt KJ, Horvath GA, Lowry N, Salomons GS, Casey B, Sinclair G, Davis C, Jakobs C, Stockler-Ipsiroglu S. Treatment of intractable epilepsy in a female with SLC6A8 deficiency. Mol Genet Metab. 2010;101:409–12. [PubMed: 20846889]
- Mercimek-Mahmutoglu S, Ndika J, Kanhai W, de Villemeur TB, Cheillan D, Christensen E, Dorison N, Hannig V, Hendriks Y, Hofstede FC, Lion-Francois L, Lund AM, Mundy H, Pitelet G, Raspall-Chaure M, Scott-Schwoerer JA, Szakszon K, Valayannopoulos V, Williams M, Salomons GS. Thirteen new patients with guanidinoacetate methyltransferase deficiency and functional characterization of nineteen novel missense variants in the GAMT gene. Hum Mutat. 2014;35:462–9. [PubMed: 24415674]
- Mercimek-Mahmutoglu S, Pop A, Kanhai W, Fernandez Ojeda M, Holwerda U, Smith D, Loeber JG, Schielen PC, Salomons GS. A pilot study to estimate incidence of guanidinoacetate methyltransferase deficiency in newborns by direct sequencing of the GAMT gene. Gene. 2016;575:127–31. [PubMed: 26319512]
- Mercimek-Mahmutoglu S, Sinclair G, van Dooren SJ, Kanhai W, Ashcraft P, Michel OJ, Nelson J, Betsalel OT, Sweetman L, Jakobs C, Salomons GS. Guanidinoacetate methyltransferase deficiency: first steps to newborn screening for a treatable neurometabolic disease. Mol Genet Metab. 2012;107:433–7. [PubMed: 23031365]
- Mhanni AA, Rockman-Greenberg C, Ryner L, Bunge M. Prenatal onset of the neuroradiologic phenotype of pyruvate carboxylase deficiency due to homozygous PC c.1828G > A mutations. JIMD Rep. 2021;61:42–7. [PMC free article: PMC8411104] [PubMed: 34485016]
- Mørkrid L, Rowe AD, Elgstoen KB, Olesen JH, Ruijter G, Hall PL, Tortorelli S, Schulze A, Kyriakopoulou L, Wamelink MM, van de Kamp JM, Salomons GS, Rinaldo P. Continuous age- and sex-adjusted reference intervals of urinary markers for cerebral creatine deficiency disorders: a novel approach to the definition of reference intervals. Clin Chem. 2015;61:760–8. [PubMed: 25759465]
- Nänto-Salonen K, Komu M, Lundbom N, Heinänen K, Alanen A, Sipilä I, Simell O. Reduced brain creatine in gyrate atrophy of the choroid and retina with hyperornithinemia. Neurology. 1999;53:303–7. [PubMed: 10430418]
- O'Rourke DJ, Ryan S, Salomons G, Jakobs C, Monavari A, King MD. Guanidinoacetate methyltransferase (GAMT) deficiency: late onset of movement disorder and preserved expressive language. Dev Med Child Neurol. 2009;51:404–7. [PubMed: 19388150]
- Pasquali M, Schwarz E, Jensen M, Yuzyuk T, DeBiase I, Randall H, Longo N. Feasibility of newborn screening for guanidinoacetate methyltransferase (GAMT) deficiency. J Inherit Metab Dis. 2014;37:231–6. [PubMed: 24276113]
- Pitt JJ, Tzanakos N, Nguyen T. Newborn screening for guanidinoacetate methyl transferase deficiency. Mol Genet Metab. 2014;111:303–4. [PubMed: 24477282]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Sinclair GB, van Karnebeek CDM, Ester M, Boyd F, Nelson T, Stockler-Ipsiroglu S, Vallance H. A three-tier algorithm for guanidinoacetate methyltransferase (GAMT) deficiency newborn screening. Mol Genet Metab. 2016;118:173–7. [PubMed: 27233226]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Stockler-Ipsiroglu S, van Karnebeek C, Longo N, Korenke GC, Mercimek-Mahmutoglu S, Marquart I, Barshop B, Grolik C, Schlune A, Angle B, Araújo HC, Coskun T, Diogo L, Geraghty M, Haliloglu G, Konstantopoulou V, Leuzzi V, Levtova A, Mackenzie J, Maranda B, Mhanni AA, Mitchell G, Morris A, Newlove T, Renaud D, Scaglia F, Valayannopoulos V, van Spronsen FJ, Verbruggen KT, Yuskiv N, Nyhan W, Schulze A. Guanidinoacetate methyltransferase (GAMT) deficiency: outcomes in 48 individuals and recommendations for diagnosis, treatment and monitoring. Mol Genet Metab. 2014;111:16–25. [PubMed: 24268530]
- Stockler-Ipsiroglu S, Apatean D, Battini R, DeBrosse S, Dessoffy K, Edvardson S, Eichler F, Johnston K, Koeller DM, Nouioua S, Tazir M, Verma A, Dowling MD, Wierenga KJ, Wierenga AM, Zhang V, Wong LJ. Arginine:glycine amidinotransferase (AGAT) deficiency: clinical features and long term outcomes in 16 patients diagnosed worldwide. Mol Genet Metab. 2015;116:252–9. [PubMed: 26490222]
- Ullio-Gamboa G, Udobi KC, Dezard S, Perna MK, Miles KN, Costa N, Taran F, Pruvost A, Benoit JP, Skelton MR, Lonlay P, Mabondzo A. Dodecyl creatine ester-loaded nanoemulsion as a promising therapy for creatine transporter deficiency. Nanomedicine (Lond). 2019;14:1579–93. [PMC free article: PMC6613044] [PubMed: 31038003]
- Valayannopoulos V, Boddaert N, Chabli A, Barbier V, Desguerre I, Philippe A, Afenjar A, Mazzuca M, Cheillan D, Munnich A, de Keyzer Y, Jakobs C, Salomons GS, de Lonlay P. Treatment by oral creatine, L-arginine and L-glycine in six severely affected patients with creatine transporter defect. J Inherit Metab Dis. 2012;35:151–7. [PubMed: 21660517]
- van de Kamp JM, Betsalel OT, Mercimek-Mahmutoglu S, Abdulhoul L, Grunewald S, Anselm I, Azzouz H, Bratkovic D, de Brouwer A, Hamel B, Kleefstra T, Yntema H, Campistol J, Vilaseca MA, Cheillan D, D'Hooghe M, Diogo L, Garcia P, Valongo C, Fonseca M, Frints S, Wilcken B, von der Haar S, Meijers-Heijboer HE, Hofstede F, Johnson D, Kant SG, Lion-Francois L, Pitelet G, Longo N, Maat-Kievit JA, Monteiro JP, Munnich A, Muntau AC, Nassogne MC, Osaka H, Ounap K, Pinard JM, Quijano-Roy S, Poggenburg I, Poplawski N, Abdul-Rahman O, Ribes A, Arias A, Yaplito-Lee J, Schulze A, Schwartz CE, Schwenger S, Soares G, Sznajer Y, Valayannopoulos V, Van Esch H, Waltz S, Wamelink MM, Pouwels PJ, Errami A, van der Knaap MS, Jakobs C, Mancini GM, Salomons GS. Phenotype and genotype in 101 males with X-linked creatine transporter deficiency. J Med Genet. 2013a;50:463–72. [PubMed: 23644449]
- van de Kamp JM, Jakobs C, Gibson KM, Salomons GS. New insights into creatine transporter deficiency: the importance of recycling creatine in the brain. J Inherit Metab Dis. 2013b;36:155–6. [PubMed: 22968583]
- van de Kamp JM, Mancini GM, Pouwels PJ, Betsalel OT, van Dooren SJ, de Koning I, Steenweg ME, Jakobs C, van der Knaap MS, Salomons GS. Clinical features and X-inactivation in females heterozygous for creatine transporter defect. Clin Genet. 2011;79:264–72. [PubMed: 20528887]
- van de Kamp JM, Mancini GM, Salomons GS. X-linked creatine transporter deficiency: clinical aspects and pathophysiology. J Inherit Metab Dis. 2014;37:715–33. [PubMed: 24789340]
- van de Kamp JM, Pouwels PJ, Aarsen FK, Ten Hoopen LW, Knol DL, de Klerk JB, de Coo IF, Huijmans JG, Jakobs C, van der Knaap MS, Salomons GS, Mancini GM. Long-term follow-up and treatment in nine boys with X-linked creatine transporter defect. J Inherit Metab Dis. 2012;35:141–9. [PMC free article: PMC3249187] [PubMed: 21556832]
- van Spronsen FJ, Reijngoud DJ, Verhoeven NM, Soorani-Lunsing RJ, Jakobs C, Sijens PE. High cerebral guanidinoacetate and variable creatine concentrations in argininosuccinate synthetase and lyase deficiency: implications for treatment. Mol Genet Metab. 2006;89:274–6. [PubMed: 16580861]
- Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev. 2000;80:1107–213. [PubMed: 10893433]
Publication Details
Author Information and Affiliations
Faculty of Medicine and Dentistry
University of Alberta
Edmonton, Alberta, Canada
Laboratory Genetic Metabolic Diseases
Amsterdam University Medical Centers
Amsterdam, the Netherlands
Publication History
Initial Posting: January 15, 2009; Last Update: February 10, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Mercimek-Andrews S, Salomons GS. Creatine Deficiency Disorders. 2009 Jan 15 [Updated 2022 Feb 10]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

