Summary
Clinical characteristics.
DYNC1H1-related disorders are primarily characterized by an axonal neuropathy with a wide phenotypic spectrum ranging from a neuromuscular-only phenotype (DYNC1H1-related neuromuscular disorder, or DYNC1H1-NMD) to phenotypes involving both the central nervous system and peripheral nervous system referred to collectively as DYNC1H1-related neurodevelopmental disorder (DYNC1H1-NDD).
DYNC1H1-NMD manifestations are limited to the peripheral nervous system and characterized predominantly by motor neuropathy initially most pronounced in the lower limbs; muscle weakness and atrophy variably associated with foot deformities, contractures, and other skeletal involvement; and/or delayed motor milestones.
DYNC1H1-NDD manifestations include motor axonal neuropathy and often global developmental delay / intellectual disability, epilepsy, neurobehavioral/psychiatric manifestations, and movement disorders with or without malformations of cortical development and/or microcephaly. In an individual with more significant central nervous system involvement, the motor axonal neuropathy may not be evident clinically and, thus, is only detected on further evaluation such as electrophysiologic testing.
Diagnosis/testing.
The diagnosis of a DYNC1H1-related disorder is established in a proband with suggestive findings and a heterozygous pathogenic variant in DYNC1H1 identified by molecular genetic testing.
Management.
Treatment of manifestations: For all individuals with a DYNC1H1-related disorder, supportive treatment typically includes multidisciplinary care by specialists in pediatric neurology, developmental pediatrics, orthopedics, physical medicine and rehabilitation, physical therapy, and medical genetics/genetic counseling. For individuals with DYNC1H1-NDD, supportive treatment often also includes care by specialists in mental health, pediatric gastroenterology, speech-language pathology, nutrition for feeding difficulties, and ophthalmology.
Surveillance: Monitoring existing manifestations, the individual's response to supportive care, and the emergence of new manifestations should be performed routinely by members of the multidisciplinary care team.
Genetic counseling.
DYNC1H1-related disorders are autosomal dominant disorders typically caused by a de novo pathogenic variant. Most individuals with DYNC1H1-NMD have the disorder as the result of a de novo pathogenic variant, although transmission of a DYNC1H1 pathogenic variant from an affected to parent to an affected child has been reported in several families. Almost all individuals diagnosed with DYNC1H1-NDD have the disorder as the result of a de novo pathogenic variant to date. Once the DYNC1H1 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
GeneReview Scope
DYNC1H1-related disorders are primarily characterized by an axonal neuropathy with a wide phenotypic spectrum ranging from a neuromuscular-only phenotype – referred to as DYNC1H1-related neuromuscular disorder (DYNC1H1-NMD) – to phenotypes involving both the central nervous system and peripheral nervous system referred to collectively as DYNC1H1-related neurodevelopmental disorder (DYNC1H1-NDD). The terms DYNC1H1-NMD and DYNC1H1-NDD encompass and replace original designations used to refer to overlapping aspects of DYNC1H1-related peripheral nervous system features (i.e., Charcot-Marie-Tooth disease and spinal muscular atrophy with lower extremity predominance) and DYNC1H1-related central nervous system features (i.e., autosomal dominant intellectual developmental disorder 13).
Table
DYNC1H1-Related Disorders
Diagnosis
Suggestive Findings
A DYNC1H1-related disorder should be considered in a proband with the following age-related clinical and electrophysiologic findings of axonal neuropathy with or without developmental delay / intellectual disability and related central nervous findings, as well as family history.
DYNC1H1-Related Neuromuscular Disorder (DYNC1H1-NMD)
Axonal neuropathy. At all ages across the phenotypic spectrum of DYNC1H1-related disorders:
- Muscle weakness and reduced-to-absent reflexes that can manifest:
- Prenatally/perinatally in fetuses/newborns with severe muscular hypotonia resulting in reduced fetal movements and contractures;
- In young children as gross motor developmental delays and positive Gower sign and muscle atrophy in those with significantly decreased strength.
Note that in individuals with primarily central nervous system involvement the signs of axonal neuropathy may be mild or masked and thus may only be detected on electrophysiologic evaluation. - Secondary skeletal involvement due to progressive muscle weakness that can manifest:
- In infants and toddlers as foot deformities, contractures, spine deformities, and/or congenital hip dysplasia/dislocation;
- In older individuals due to spastic quadri- or paraparesis.
- Spastic paraparesis first involves the lower limbs, manifesting as gait abnormalities and ataxia progressing to spastic quadriparesis and akinesia. Upper limb involvement is less pronounced at disease onset but may progress (over ten to 20 years) during adolescence or adulthood.
Sensory involvement, especially of the lower limbs, may be consistent with a sensory neuropathy.
Electrophysiologic findings
- Nerve conduction studies (NCS) may reveal low/decreased motor amplitude [Beecroft et al 2017, Chen et al 2017], decreased velocity [Strickland et al 2015], and/or single, small compound muscle action potential amplitudes as part of the axonal neuropathy [Harms et al 2010].
- Electromyography (EMG) may reveal chronic, axonal neurogenic changes comprising denervation and faulty reinnervation, large-amplitude and long-duration motor unit potentials, positive sharp waves, giant potentials during slight contraction, and neurogenic recruitment patterns [Harms et al 2010, Niu et al 2015, Scoto et al 2015, Ding et al 2016, Das et al 2018, Fernández Perrone et al 2022, Li et al 2022].
- NCS and EMG may be consistent with sensory neuropathy [Weedon et al 2011].
Muscle MRI shows atrophy with fatty replacement/infiltration and compensatory hypertrophy in lower limb muscles, mostly the quadriceps.
DYNC1H1-Related Neurodevelopmental Disorder (DYNC1H1-NDD)
In addition to axonal neuropathy described above, all individuals with DYNC1H1-NDD have neurodevelopmental delay with varying degree of (gross or fine) motor, speech, and/or cognitive delay.
Additionally, some individuals manifest the following:
- Epilepsy, typically infantile onset; multiple seizure types can occur, including infantile epileptic spasms syndrome and focal and generalized motor seizures (tonic, myoclonic, and tonic-clonic).
- Neurobehavioral/psychiatric manifestations, most commonly attention-deficit hyperactivity disorder and/or autism spectrum disorder.
- Movement disorders, typically later onset, including resting, intention, or generalized tremor, spastic grasp, and parkinsonism
Brain MRI abnormalities that may or may not be present comprise a spectrum of severity, including the following:
- Malformations of cortical development including pachygyria, lissencephaly, polymicrogyria, and other types of cortical dysgyria
- Dysgenesis or agenesis of the corpus callosum
- Enlarged ventricles
- Gray matter heterotopia
- Pontocerebellar hypoplasia
- White matter abnormalities (less common)
Family History
Because DYNC1H1-related disorders are typically caused by a de novo pathogenic variant, most probands represent a simplex case (i.e., a single occurrence in a family). Rarely, in families with neuromuscular-predominant phenotypes, the family history may be consistent with autosomal dominant inheritance (e.g., affected males and females in multiple generations).
Establishing the Diagnosis
The diagnosis of a DYNC1H1-related disorder is established in a proband with suggestive findings and a heterozygous pathogenic (or likely pathogenic) variant in DYNC1H1 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of a heterozygous DYNC1H1 variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing). Gene-targeted testing requires that the clinician determine which gene(s) are likely involved (see Option 1), whereas comprehensive genomic testing does not (see Option 2).
Note: Single-gene testing (sequence analysis of DYNC1H1, followed by gene-targeted deletion/duplication analysis) is rarely useful and typically NOT recommended.
Option 1
A multigene panel that includes DYNC1H1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in DYNC1H1-Related Disorders
Clinical Characteristics
Clinical Description
DYNC1H1-related disorders are primarily characterized by an axonal neuropathy with a wide phenotypic spectrum, ranging from a neuromuscular-only phenotype (DYNC1H1-related neuromuscular disorder) to phenotypes involving both the central nervous system and peripheral nervous system referred to collectively as DYNC1H1-related neurodevelopmental disorder [Amabile et al 2020, Becker et al 2020, Dafsari et al 2021].
DYNC1H1-related neuromuscular disorder (DYNC1H1-NMD). Manifestations are limited to the peripheral nervous system (PNS) and characterized predominantly by motor neuropathy, initially most pronounced in the lower limbs; muscle weakness and atrophy variably associated with foot deformities, contractures, and other skeletal involvement; and/or delayed motor milestones.
DYNC1H1-related neurodevelopmental disorder (DYNC1H1-NDD). Manifestations include motor axonal neuropathy and global developmental delay / intellectual disability, epilepsy, neurobehavioral/psychiatric manifestations, and movement disorders with or without malformations of cortical development (MCD) and or microcephaly. In an individual with more significant central nervous system (CNS) involvement, the motor axonal neuropathy may not be evident clinically and thus only detected on further evaluation such as electrophysiologic testing.
To date, more than 200 individuals with DYNC1H1-related disorders have been identified [Amabile et al 2020, Becker et al 2020]. The following comparison of the two phenotypic designations for DYNC1H1-related disorders is based on these reports (see Figure 1 and Table 2).
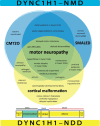
Table 2.
DYNC1H1-Related Disorders: Comparison of Presentations at Initial Evaluation
DYNC1H1-Related Neuromuscular Disorder (DYNC1H1-NMD)
Motor axonal neuropathy. In general, the proximal muscles of the lower limbs initially are most severely affected, with progression during childhood to the entire proximal lower limbs and subsequently during adolescence or adulthood to the upper limbs [Harms et al 2010, Tsurusaki et al 2012, Peeters et al 2015, Scoto et al 2015, Ding et al 2016, Hertecant et al 2016]. Up to 95% of individuals have reduced lower limb muscle strength and about 20% have reduced upper limb strength [Becker et al 2020].
Initial findings of lower limb involvement include decreased muscle tone and weakness due to muscle atrophy and reduced muscle mass. In some infants, decreased fetal movements result in secondary contractures evident at birth [Scoto et al 2015, Becker et al 2020], including the following:
- Foot deformities, present in around 50% of individuals, such as pes cavus (in around half of all cases), pes equinus, pes equinovarus, pes adductus, talus verticalis, shortened forefoot, slender/hammer toes, pes calcaneus, hyperextension deformities, and bilateral foot drop [Weedon et al 2011, Beecroft et al 2017, Chen et al 2017, Chan et al 2018, Becker et al 2020, Liu et al 2023]
- Other contractures, present in around 10% of individuals, almost exclusively affecting the lower limb, including the hips, iliotibial ligament, knees, and Achilles tendon; can also involve the upper limb (especially the thumbs) [Scoto et al 2015, Amabile et al 2020, Becker et al 2020]. Congenital unilateral or bilateral hip dysplasia and/or dislocation, a relatively rare manifestation, may be evident prenatally on ultrasound examination or after birth [Scoto et al 2015].
Sensory involvement, especially of the lower limbs, can include transient paresthesias, neuropathic pain, reduced or lost proprioception, and reduced response to pinprick, fine touch, and/or vibration. Age-related progression may manifest during adulthood as leg fatigue and pain [Harms et al 2010].
Motor development is moderately to severely delayed in about 50% of individuals with DYNC1H1-NMD [Becker et al 2020, Dafsari et al 2021]. An abnormal waddling gait, present in 30% of individuals due to reduced proprioception and muscle weakness in the proximal lower limbs, is characterized by minor imbalance or recurrent falls and difficulties in running [Weedon et al 2011, Niu et al 2015, Scoto et al 2015, Becker et al 2020].
As neuropathy progresses, the gait may become ataxic and walking aids such as canes or wheelchairs may be required [Chan et al 2018].
Other less common motor involvement includes the following:
- Difficulty feeding and poor weight gain due to orofacial hypotonia [Gelineau-Morel et al 2016]
- Rare instances of weakness in the periscapular or extraocular muscles [Weedon et al 2011, Chan et al 2018, Amabile et al 2020]
Histological findings. Click here (pdf) for histologic findings in PNS studies.
DYNC1H1-Related Neurodevelopmental Disorder (DYNC1H1-NDD)
Individuals with DYNC1H1-NDD exhibit the motor axonal neuropathy described previously for DYNC1H1-NMD, as well as additional variable features.
Neurodevelopmental delays including motor, speech, and/or cognitive development may occur [Weedon et al 2011, Becker et al 2020]. Developmental regression in these three domains has been reported in a few individuals [Amabile et al 2020, Yang et al 2021].
- Motor development is moderately to severely delayed in approximately 50% of individuals [Becker et al 2020]. There may be several pathomechanisms underlying motor developmental delay that may include axonal neuropathy in the PNS or CNS and/or malformations of cortical development.Note: Delay of gross motor milestones that manifests in the legs only (e.g., walking) may suggest PNS involvement (i.e., motor axonal neuropathy) in a child with DYNC1H1-NMD, which usually presents initially with lower extremity weakness. Upper extremity involvement was only reported in rare instances [Scoto et al 2015].
- Speech development. Impaired speech development may be present as part of a global neurodevelopmental disorder in those with intellectual disability (ID) in DYNC1H1-NDD.
- Cognitive development is delayed in around 40% of individuals. The severity ranges from mild learning problems to severe ID. While there is no clear correlation between brain MRI findings and the degree of ID, presumably children with severe malformations of cortical development are likely to have more severe developmental delays [Becker et al 2020].
Neurobehavioral/psychiatric manifestations affect more than 15% of individuals with DYNC1H1-NDD [Becker et al 2020]. The most common is attention-deficit hyperactivity disorder. Other behavioral disorders include attention disorders without hyperactivity, dyslexia, autism spectrum disorder, aggressive behavior, and motor stereotypies [Chan et al 2018, Amabile et al 2020, Becker et al 2020].
Epilepsy. Seizures, reported in nearly 40% of individuals, are infantile onset in about 60% of all individuals with seizures [Becker et al 2020, Liu et al 2023]. The most common seizure types are focal (27%), followed by generalized onset (10%) and mixed focal and generalized onset (8%) [Chung et al 2022]. Infantile epileptic spasms syndrome (IESS) was reported in over 10% of individuals with seizures [Yang et al 2021, Su et al 2022, Liu et al 2023].
Other reported specific electroclinical syndromes are centrotemporal epilepsy, acquired aphasia syndrome, focal epilepsy of structural origin, and Lennox-Gastaut syndrome [Liu et al 2023].
Electroencephalography (EEG) may show different patterns according to seizure semiology, including the following:
- Hypsarrhythmia and high-amplitude ictal rhythmic waves in individuals with IESS. With time hypsarrhythmia can evolve into an electrographic pattern characterized by focal or multifocal epileptiform discharges [Yang et al 2021, Su et al 2022].
- Focal or multifocal epileptiform discharges in focal-onset seizures [Matsumoto et al 2021, Ji et al 2022, Liu et al 2023]
- Generalized spike-wave complexes, irregular polyspikes, and slow waves, mixed theta and delta frequencies, and periodic spike and slow wave activity in generalized seizures [Yang et al 2021, Liu et al 2023]
- Slow background activity in most affected individuals
Movement disorders. Parkinsonism, reported in one individual with global developmental delay, extrapyramidal findings (bradykinesia, hypokinesia, cogwheel rigidity, small step walking, difficulty in initiating movements, hypomimic face, and a resting tremor), responded favorably to treatment with levodopa [Szczałuba et al 2018].
Limb ataxia has been rarely reported [Strickland et al 2015, Fernández Perrone et al 2022].
Vocal cord paresis and dysarthria have been rarely reported and may be different from the speech disorders associated with neurodevelopmental delay [Jamuar et al 2014, Zillhardt et al 2016, Amabile et al 2020].
Brain MRI abnormalities have been detected on in more than 60% of individuals studied and include the following [Poirier et al 2013, Fiorillo et al 2014, Jamuar et al 2014, Scoto et al 2015, Gelineau-Morel et al 2016, Hertecant et al 2016, Su et al 2022, Liu et al 2023]:
- Malformations of cortical development including pachygyria (that may resemble lissencephaly), polymicrogyria, a much broader range of cortical dysgyria (48%), and dysgenesis or agenesis of the corpus callosum (23%)
- Ventriculomegaly (15%)
- Cerebellar hypoplasia (14%)
- Gray matter heterotopia (11%)
- Brain stem hypoplasia (9%)
Other findings seen in one or a few individuals include abnormalities of the white matter [Gelineau-Morel et al 2016, Chan et al 2018, Liu et al 2023], cortical atrophy [Becker et al 2020], dysmorphic basal ganglia [Poirier et al 2013], schizencephaly [Liu et al 2023], arachnoid cyst [Chan et al 2018], large cerebrospinal fluid spaces [Hertecant et al 2016], and hydrocephalus [Scoto et al 2015].
Microcephaly, affecting about 5% of individuals, is most often congenital [Poirier et al 2013, Laquerriere et al 2017]. In other individuals, head circumference may be normal at birth, with microcephaly becoming evident postnatally [Hertecant et al 2016, Laquerriere et al 2017, Becker et al 2020].
Neuropathologic examination. Click here (pdf) for findings from neuropathologic examinations.
Other Findings
The following findings have been observed across the entire phenotypic spectrum of DYNC1H1-related disorders:
- Ophthalmologic findings, reported in about 10% of individuals, most frequently bilateral cataracts [Amabile et al 2020]. Other findings include amblyopia, strabismus, and visual impairment [Weedon et al 2011, Gelineau-Morel et al 2016, Chen et al 2017, Becker et al 2020].
- Gastrointestinal manifestations, reported in <5% of individuals, including gastric volvulus; incontinence; constipation; dysmotility of the stomach, small bowel, and descending and sigmoid colon; postprandial hypomobility; and omphalocele [Strickland et al 2015, Gelineau-Morel et al 2016, Chan et al 2018, Amabile et al 2020]
- Congenital anomalies of kidney and urinary tract (CAKUT), reported in <5% of individuals, including dilatated uropathy [Becker et al 2020]
- Other manifestations, including intermittent painful muscle cramps with moderate exercise [Beecroft et al 2017], intrauterine growth restriction, hydrops fetalis [Zillhardt et al 2016], osteocutaneous anomalies (e.g., prominent calcanei, cutis laxa), accessory spleen, congenital anterior diaphragmatic hernia, syringomyelia [Becker et al 2020], cryptorchidism [Minardi et al 2020], hypospadias [Amabile et al 2020], celiac disease, and eosinophilic esophagitis [Fernández Perrone et al 2022].
- Cardiovascular manifestations, perhaps incidental due to their prevalence in the general population, were reported in <5% of DYNC1H1-related disorders and include mild aortic valve insufficiency, bicuspid aortic valve, anomalous pulmonary venous drainage, and atrial septum defect resulting in ventricular hypertrophy [Amabile et al 2020, Becker et al 2020, Liu et al 2023].
Genotype-Phenotype Correlations
Several genotype-phenotype correlations have been observed based on the location of pathogenic variants within the four main functional domains of DYNC1H1 [Becker et al 2020, Dafsari et al 2021] (see Figure 1); however, limited data to date prevent making more precise determinations.
Beginning tail (comprising amino acids 1-299 and 1141-1373). Pathogenic variants in this region are mainly associated with DYNC1H1-NDD and behavioral abnormalities.
Dimerization domain (comprising amino acids 300-1140). Pathogenic variants in this region lead to generally milder, primarily neuromuscular disorders (DYNC1H1-NMD).
Linker domain (comprising amino acids 1374-1867). Pathogenic variants in this region are particularly associated with DYNC1H1-NDD and behavioral abnormalities.
Motor domain (comprising amino acids 1868-4221). Pathogenic variants in this domain have been found mainly in individuals with DYNC1H1-NDD with malformations of cortical development, epilepsy, and neurobehavioral/psychiatric manifestations [Amabile et al 2020, Becker et al 2020, Chung et al 2022].
Penetrance
Penetrance in previously reported families with known recurrences is high for DYNC1H1-related disorders [Schiavo et al 2013].
It is assumed that penetrance is age-related because of observed disease progression over time [Dafsari et al 2021].
Nomenclature
Phenotypes associated with DYNC1H1 pathogenic variants have been classified into distinct clinical entities. Because these entities have overlapping and often mixed phenotypes, a new holistic classification has been proposed by Amabile et al [2020], Becker et al [2020], and Dafsari et al [2021] (see Table 3).
Table 3.
DYNC1H1-Related Disorders: Holistic Classification and Original Designations
Prevalence
The birth prevalence for DYNC1H1-related disorders is unknown but expected to be low. To date more than 200 individuals have been reported with DYNC1H1-related disorders (see Clinical Description).
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline heterozygous DYNC1H1 pathogenic variants.
Sporadic tumors (including primary gallbladder cancer, colorectal cancer, and drug-resistant gastric cancer) occurring as single tumors in the absence of any other findings of DYNC1H1-related disorders frequently contain a somatic pathogenic variant in DYNC1H1 (mostly localized in the DYNC1H1 motor domain [Sucularli & Arslantas 2017, Pan et al 2021]) that is not present in the germline. In these circumstances predisposition to these tumors is not heritable.
Differential Diagnosis
Because DYNC1H1-related disorders are clinically indistinguishable from many other inherited disorders with similar neurologic findings, diagnosing this condition on clinical grounds only without molecular genetic testing is not feasible. All disorders with neuromuscular and/or neurodevelopmental features without other distinctive findings should be considered in the differential diagnosis. See:
- OMIM Phenotypic Series:
Specific disorders with a high degree of clinical overlap include those listed in Table 4a (DYNC1H1-related neuromuscular disorder) and Table 4b (DYNC1H1-related neurodevelopmental disorder).
Table 4a.
DYNC1H1-Related Neuromuscular Disorder: Selected Disorders in the Differential Diagnosis
Table 4b.
DYNC1H1-Related Neurodevelopmental Disorder: Selected Disorders in the Differential Diagnosis
Management
No clinical practice guidelines for DYNC1H1-related disorders have been published. In the absence of published guidelines, the following recommendations are based on the authors' personal experience managing individuals with this disorder.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with a DYNC1H1-related disorder, the evaluations summarized in Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 5.
DYNC1H1-Related Disorders: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
There is no cure for DYNC1H1-related disorders. Supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in neurology, physiatry, orthopedics, and physical and occupational therapy (see Table 6).
Table 6.
DYNC1H1-Related Disorders: Treatment of Manifestations
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
- For muscle tone abnormalities and movement disorders including hypertonia or dystonia, consider involving appropriate specialists to aid in management of baclofen, tizanidine, botulinum toxin, anti-parkinsonian medications, or orthopedic procedures.
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Neurobehavioral/psychiatric manifestations. Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder, when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 7 are recommended.
Table 7.
DYNC1H1-Related Disorders: Recommended Surveillance
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
DYNC1H1-related disorders are autosomal dominant disorders typically caused by a de novo pathogenic variant.
- Most individuals with DYNC1H1-related neuromuscular disorder (DYNC1H1-NMD) have the disorder as the result of a de novo pathogenic variant, although transmission of a DYNC1H1 pathogenic variant from an affected parent to an affected child has been reported in several families.
- Almost all individuals diagnosed to date with DYNC1H1-related neurodevelopmental disorder (DYNC1H1-NDD) have the disorder as the result of a de novo pathogenic variant.
Risk to Family Members
Parents of a proband
- Most individuals diagnosed with a DYNC1H1-related disorder have the disorder as the result of a de novo pathogenic variant.
- Very few individuals diagnosed with a DYNC1H1-related disorder have an affected parent.
- Several large families with DYNC1H1-NMD have been reported [Beecroft et al 2017].
- Transmission of a DYNC1H1 pathogenic variant from a parent with malformations of cortical development and epilepsy to two affected children has been reported in one family [Poirier et al 2013, Chung et al 2022].
- If the proband appears to be the only affected family member (i.e., a simplex case), molecular genetic testing is recommended for the parents of the proband to evaluate their genetic status and to inform recurrence risk assessment.
- If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism [Zillhardt et al 2016].* Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ (gonadal) cells only.* A parent with somatic and germline mosaicism for a DYNC1H1 pathogenic variant may be mildly/minimally affected.
- The family history of some individuals diagnosed with DYNC1H1-NMD may appear to be negative because of failure to recognize the disorder in family members or age-related reduced penetrance. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has demonstrated that neither parent is heterozygous for the pathogenic variant identified in the proband.
Sibs of a proband. The risk to the sibs of the proband depends on the clinical/genetic status of the proband's parents:
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%.
- If the DYNC1H1 pathogenic variant identified in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental germline mosaicism (maternal germline mosaicism was reported in a family in which two fetuses were found to have malformations of cortical development) [Zillhardt et al 2016].
- If the parents have not been tested for the DYNC1H1 pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for DYNC1H1-NMD because of the possibility of parental germline mosaicism.
Offspring of a proband
- Each child of an individual with DYNC1H1-NMD has a 50% chance of inheriting the DYNC1H1 pathogenic variant.
- Individuals with severe DYNC1H1-NDD are not known to reproduce; however, many are not yet of reproductive age.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the DYNC1H1 pathogenic variant, the parent's family members may be at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the DYNC1H1 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal and preimplantation genetic testing. While most centers would consider use of prenatal and preimplantation genetic testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- DYNC1H1 AssociationEmail: contact@dync1h1.org
- American Association on Intellectual and Developmental Disabilities (AAIDD)Phone: 202-387-1968
- CDC - Child DevelopmentPhone: 800-232-4636
- Charcot-Marie-Tooth Association (CMTA)Phone: 800-606-2682 (toll-free); 610-427-2971Email: info@cmtausa.org
- CMT Research FoundationPhone: 404-806-7180Email: info@cmtrf.org
- European Neuromuscular Centre (ENMC)NetherlandsPhone: 31 35 5480481Email: enmc@enmc.org
- Hereditary Neuropathy FoundationPhone: 855-435-7268 (toll-free); 212-722-8396Fax: 917-591-2758Email: info@hnf-cure.org
- MedlinePlus
- VOR: Speaking out for people with intellectual and developmental disabilitiesPhone: 877-399-4867Email: info@vor.net
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
DYNC1H1-Related Disorders: Genes and Databases
Table B.
OMIM Entries for DYNC1H1-Related Disorders (View All in OMIM)
Molecular Pathogenesis
DYNC1H1 encodes cytoplasmic dynein 1 heavy chain 1, a large protein that is a crucial component of the intracellular motor complex for vesicular trafficking along microtubules vital for retrograde axonal transport in neurons. Cytoplasmic dynein 1 heavy chain 1 also plays a role in the regulation and maintenance of the Golgi apparatus, spindle pole organization, nuclear migration and positioning, and recruitment of other dynein subunits [Schiavo et al 2013, Amabile et al 2020, Li et al 2022].
The cytoplasmic dynein 1 heavy chain 1 complex comprises a core of dimerized heavy chains [Hoang et al 2017]. The C-terminal region (amino acid residues 1846-4646) is the motor domain complex; it is arranged as a heptameric ring with seven AAA domains and a stalk from which protrudes a microtubule-binding domain. While the precise pathomechanisms of DYNC1H1-related disorders remain largely elusive, several genotype-phenotype correlations have been observed based on the location of pathogenic variants within the four main functional domains of the gene [Becker et al 2020, Dafsari et al 2021] (see Genotype-Phenotype Correlation).
Mechanism of disease causation. Preliminary data available to date suggest the mechanism of disease causation is loss of function.
Variants of uncertain significance. To evaluate variants of uncertain significance in a child with an unknown phenotype, nerve conduction studies or a nerve and/or muscle biopsy may support diagnosis of peripheral nerve involvement in DYNC1H1-related neuromuscular disorder.
Chapter Notes
Author Notes
Birk Möller and Hormos Dafsari are actively involved in clinical research regarding individuals with DYNC1H1-related disorders. They would be happy to communicate with persons who have any questions regarding diagnosis of DYNC1H1-related disorders or other considerations.
Hormos Dafsari and Heinz Jungbluth are also interested in hearing from clinicians treating families affected by innate errors of autophagy and intracellular trafficking in whom no causative variant has been identified through molecular genetic testing of the genes known to be involved in this group of disorders.
Contact Hormos Dafsari to inquire about review of DYNC1H1 variants of uncertain significance.
Acknowledgments
HSD was supported by the German Society for Muscle Diseases (DGM, Da3/1), Cologne Clinician Scientist Program / Medical Faculty / University of Cologne and German Research Foundation (CCSP, DFG project no. 413543196).
HJ was supported by Action Medical Research, United Kingdom, the GOSH Charity / Sparks, and a European Commission H2020-MSCA-ITN-2017 grant.
Revision History
- 21 March 2024 (bp) Review posted live
- 21 February 2023 (hsd) Original submission
References
Literature Cited
- Amabile S, Jeffries L, McGrath JM, Ji W, Spencer-Manzon M, Zhang H, Lakhani SA. DYNC1H1-related disorders: a description of four new unrelated patients and a comprehensive review of previously reported variants. Am J Med Genet A. 2020;182:2049-57. [PubMed: 32656949]
- Becker LL, Dafsari HS, Schallner J, Abdin D, Seifert M, Petit F, Smol T, Bok L, Rodan L, Krapels I, Spranger S, Weschke B, Johnson K, Straub V, Kaindl AM, Di Donato N, von der Hagen M, Cirak S. The clinical-phenotype continuum in DYNC1H1-related disorders-genomic profiling and proposal for a novel classification. J Hum Genet. 2020;65:1003-17. [PMC free article: PMC7719554] [PubMed: 32788638]
- Beecroft SJ, McLean CA, Delatycki MB, Koshy K, Yiu E, Haliloglu G, Orhan D, Lamont PJ, Davis MR, Laing NG, Ravenscroft G. Expanding the phenotypic spectrum associated with mutations of DYNC1H1. Neuromuscul Disord. 2017;27:607-15. [PubMed: 28554554]
- Chan SHS, van Alfen N, Thuestad IJ, Ip J, Chan AO, Mak C, Chung BH, Verrips A, Kamsteeg EJ. A recurrent de novo DYNC1H1 tail domain mutation causes spinal muscular atrophy with lower extremity predominance, learning difficulties and mild brain abnormality. Neuromuscul Disord. 2018;28:750-6. [PubMed: 30122514]
- Chen Y, Xu Y, Li G, Li N, Yu T, Yao RE, Wang X, Shen Y, Wang J. Exome sequencing identifies de novo DYNC1H1 mutations associated with distal spinal muscular atrophy and malformations of cortical development. J Child Neurol. 2017;32:379-86. [PubMed: 28193117]
- Chung CT, Lee NC, Fan SP, Hung MZ, Lin YH, Chen CH, Jao T. DYNC1H1 variant associated with epilepsy: expanding the phenotypic spectrum. Epilepsy Behav Rep. 2022;21:100580. [PMC free article: PMC9829698] [PubMed: 36636459]
- Dafsari HS, Becker LL, von der Hagen M, Cirak S. Genomic profiling in neuronal dyneinopathies and updated classifications. Am J Med Genet A. 2021;185:2607-10. [PubMed: 33991169]
- Das J, Lilleker JB, Jabbal K, Ealing J. A missense mutation in DYNC1H1 gene causing spinal muscular atrophy - lower extremity, dominant. Neurol Neurochir Pol. 2018;52:293-7. [PubMed: 29306600]
- Ding D, Chen Z, Li K, Long Z, Ye W, Tang Z, Xia K, Qiu R, Tang B, Jiang H. Identification of a de novo DYNC1H1 mutation via WES according to published guidelines. Sci Rep. 2016;6:20423. [PMC free article: PMC4742772] [PubMed: 26846447]
- Fernández Perrone AL, Moreno Fernández P, Álvarez S, Fernández-Jaén A. DYNC1H1 de novo mutation, spinal muscular atrophy and attention problems. Neurologia (Engl Ed). 2022;37:406-9. [PubMed: 35606327]
- Fiorillo C, Moro F, Yi J, Weil S, Brisca G, Astrea G, Severino M, Romano A, Battini R, Rossi A, Minetti C, Bruno C, Santorelli FM, Vallee R. Novel dynein DYNC1H1 neck and motor domain mutations link distal spinal muscular atrophy and abnormal cortical development. Hum Mutat. 2014;35:298-302. [PMC free article: PMC4109683] [PubMed: 24307404]
- Gelineau-Morel R, Lukacs M, Weaver KN, Hufnagel RB, Gilbert DL, Stottmann RW. Congenital cataracts and gut dysmotility in a DYNC1H1 dyneinopathy patient. Genes (Basel). 2016;7:85. [PMC free article: PMC5083924] [PubMed: 27754416]
- Harms MB, Allred P, Gardner R Jr, Fernandes Filho JA, Florence J, Pestronk A, Al-Lozi M, Baloh RH. Dominant spinal muscular atrophy with lower extremity predominance: linkage to 14q32. Neurology. 2010;75:539-46. [PMC free article: PMC2918478] [PubMed: 20697106]
- Hertecant J, Komara M, Nagi A, Suleiman J, Al-Gazali L, Ali BR. A novel de novo mutation in DYNC1H1 gene underlying malformation of cortical development and cataract. Meta Gene. 2016;9:124-7. [PMC free article: PMC4908276] [PubMed: 27331017]
- Hoang HT, Schlager MA, Carter AP, Bullock SL. DYNC1H1 mutations associated with neurological diseases compromise processivity of dynein-dynactin-cargo adaptor complexes. Proc Natl Acad Sci U S A. 2017;114:E1597-E1606. [PMC free article: PMC5338514] [PubMed: 28196890]
- Jamuar SS, Lam AT, Kircher M, D'Gama AM, Wang J, Barry BJ, Zhang X, Hill RS, Partlow JN, Rozzo A, Servattalab S, Mehta BK, Topcu M, Amrom D, Andermann E, Dan B, Parrini E, Guerrini R, Scheffer IE, Berkovic SF, Leventer RJ, Shen Y, Wu BL, Barkovich AJ, Sahin M, Chang BS, Bamshad M, Nickerson DA, Shendure J, Poduri A, Yu TW, Walsh CA. Somatic mutations in cerebral cortical malformations. N Engl J Med. 2014;371:733-43. [PMC free article: PMC4274952] [PubMed: 25140959]
- Ji C, Wu D, Wang K. Whole-exome sequencing identifies a novel de novo variant in DYNC1H in a patient with intractable epilepsy. Neurol Sci. 2022;43:2853-8. [PubMed: 35088241]
- Kramarz C, Rossor AM. Neurological update: hereditary neuropathies. J Neurol. 2022;269:5187-91. [PMC free article: PMC9363318] [PubMed: 35596796]
- Laquerriere A, Maillard C, Cavallin M, Chapon F, Marguet F, Molin A, Sigaudy S, Blouet M, Benoist G, Fernandez C, Poirier K, Chelly J, Thomas S, Bahi-Buisson N. Neuropathological hallmarks of brain malformations in extreme phenotypes related to DYNC1H1 mutations. J Neuropathol Exp Neurol. 2017;76:195-205. [PubMed: 28395088]
- Li JT, Dong SQ, Zhu DQ, Yang WB, Qian T, Liu XN, Chen XJ. Expanding the phenotypic and genetic spectrum of neuromuscular diseases caused by DYNC1H1 mutations. Front Neurol. 2022;13:943324. [PMC free article: PMC9309508] [PubMed: 35899263]
- Liu W, Cheng M, Zhu Y, Chen Y, Yang Y, Chen H, Niu X, Tian X, Yang X, Zhang Y. DYNC1H1-related epilepsy: genotype-phenotype correlation. Dev Med Child Neurol. 2023;65:534-43. [PubMed: 36175372]
- Marchionni E, Agolini E, Mastromoro G, Guadagnolo D, Coppola G, Roggini M, Riminucci M, Novelli A, Giancotti A, Corsi A, Pizzuti A. Fetal early motor neuron disruption and prenatal molecular diagnosis in a severe BICD2-opathy. Am J Med Genet A. 2021;185:1509-14. [PubMed: 33547725]
- Matsumoto A, Kojima K, Miya F, Miyauchi A, Watanabe K, Iwamoto S, Kawai K, Kato M, Takahashi Y, Yamagata T. Two cases of DYNC1H1 mutations with intractable epilepsy. Brain Dev. 2021;43:857-62. [PubMed: 34092403]
- Minardi R, Licchetta L, Baroni MC, Pippucci T, Stipa C, Mostacci B, Severi G, Toni F, Bergonzini L, Carelli V, Seri M, Tinuper P, Bisulli F. Whole-exome sequencing in adult patients with developmental and epileptic encephalopathy: it is never too late. Clin Genet. 2020;98:477-85. [PubMed: 32725632]
- Niu Q, Wang X, Shi M, Jin Q. A novel DYNC1H1 mutation causing spinal muscular atrophy with lower extremity predominance. Neurol Genet. 2015;1:e20. [PMC free article: PMC4807905] [PubMed: 27066557]
- Pan H, Chai W, Liu X, Yu T, Sun L, Yan M. DYNC1H1 regulates NSCLC cell growth and metastasis by IFN-γ-JAK-STAT signaling and is associated with an aberrant immune response. Exp Cell Res. 2021;409:112897. [PubMed: 34717919]
- Peeters K, Bervoets S, Chamova T, Litvinenko I, De Vriendt E, Bichev S, Kancheva D, Mitev V, Kennerson M, Timmerman V, De Jonghe P, Tournev I, MacMillan J, Jordanova A. Novel mutations in the DYNC1H1 tail domain refine the genetic and clinical spectrum of dyneinopathies. Hum Mutat. 2015;36:287-91. [PubMed: 25512093]
- Poirier K, Lebrun N, Broix L, Tian G, Saillour Y, Boscheron C, Parrini E, Valence S, Pierre BS, Oger M, Lacombe D, Geneviève D, Fontana E, Darra F, Cances C, Barth M, Bonneau D, Bernadina BD, N'guyen S, Gitiaux C, Parent P, des Portes V, Pedespan JM, Legrez V, Castelnau-Ptakine L, Nitschke P, Hieu T, Masson C, Zelenika D, Andrieux A, Francis F, Guerrini R, Cowan NJ, Bahi-Buisson N, Chelly J. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet. 2013;45:639-47. [PMC free article: PMC3826256] [PubMed: 23603762]
- Schiavo G, Greensmith L, Hafezparast M, Fisher EM. Cytoplasmic dynein heavy chain: the servant of many masters. Trends Neurosci. 2013;36:641-51. [PMC free article: PMC3824068] [PubMed: 24035135]
- Scoto M, Rossor AM, Harms MB, Cirak S, Calissano M, Robb S, Manzur AY, Martínez Arroyo A, Rodriguez Sanz A, Mansour S, Fallon P, Hadjikoumi I, Klein A, Yang M, De Visser M, Overweg-Plandsoen WC, Baas F, Taylor JP, Benatar M, Connolly AM, Al-Lozi MT, Nixon J, de Goede CG, Foley AR, Mcwilliam C, Pitt M, Sewry C, Phadke R, Hafezparast M, Chong WK, Mercuri E, Baloh RH, Reilly MM, Muntoni F. Novel mutations expand the clinical spectrum of DYNC1H1-associated spinal muscular atrophy. Neurology. 2015;84:668-79. [PMC free article: PMC4336105] [PubMed: 25609763]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197-207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Strickland AV, Schabhüttl M, Offenbacher H, Synofzik M, Hauser NS, Brunner-Krainz M, Gruber-Sedlmayr U, Moore SA, Windhager R, Bender B, Harms M, Klebe S, Young P, Kennerson M, Garcia AS, Gonzalez MA, Züchner S, Schule R, Shy ME, Auer-Grumbach M. Mutation screen reveals novel variants and expands the phenotypes associated with DYNC1H1. J Neurol. 2015;262:2124-34. [PMC free article: PMC4573829] [PubMed: 26100331]
- Su T, Yan Y, Hu Q, Liu Y, Xu S. De novo DYNC1H1 mutation causes infantile developmental and epileptic encephalopathy with brain malformations. Mol Genet Genomic Med. 2022;10:e1874. [PMC free article: PMC8922968] [PubMed: 35099838]
- Sucularli C, Arslantas M. Computational prediction and analysis of deleterious cancer associated missense mutations in DYNC1H1. Mol Cell Probes. 2017;34:21-29. [PubMed: 28455235]
- Szczałuba K, Szymańska K, Rydzanicz M, Ciara E, Walczak A, Piekutowska-Abramczuk D, Kosińska J, Jacoszek A, Czerska K, Biernacka A, Laure-Kamionowska M, Gasperowicz P, Pronicka E, Płoski R. A de novo loss-of-function DYNC1H1 mutation in a patient with parkinsonian features and a favourable response to levodopa. Clin Genet. 2018;93:1107-8. [PubMed: 29243232]
- Tsurusaki Y, Saitoh S, Tomizawa K, Sudo A, Asahina N, Shiraishi H, Ito J, Tanaka H, Doi H, Saitsu H, Miyake N, Matsumoto N. A DYNC1H1 mutation causes a dominant spinal muscular atrophy with lower extremity predominance. Neurogenetics. 2012;13:327-32. [PubMed: 22847149]
- Weedon MN, Hastings R, Caswell R, Xie W, Paszkiewicz K, Antoniadi T, Williams M, King C, Greenhalgh L, Newbury-Ecob R, Ellard S. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet. 2011;89:308-12. [PMC free article: PMC3155164] [PubMed: 21820100]
- Yang H, Gong P, Jiao X, Niu Y, Zhou Q, Zhang Y, Yang Z. De novo variants in the DYNC1H1 gene associated with infantile spasms. Front Neurol. 2021;12:733178. [PMC free article: PMC8603382] [PubMed: 34803881]
- Zillhardt JL, Poirier K, Broix L, Lebrun N, Elmorjani A, Martinovic J, Saillour Y, Muraca G, Nectoux J, Bessieres B, Fallet-Bianco C, Lyonnet S, Dulac O, Odent S, Rejeb I, Ben Jemaa L, Rivier F, Pinson L, Geneviève D, Musizzano Y, Bigi N, Leboucq N, Giuliano F, Philip N, Vilain C, Van Bogaert P, Maurey H, Beldjord C, Artiguenave F, Boland A, Olaso R, Masson C, Nitschké P, Deleuze JF, Bahi-Buisson N, Chelly J. Mosaic parental germline mutations causing recurrent forms of malformations of cortical development. Eur J Hum Genet. 2016;24:611-4. [PMC free article: PMC4929884] [PubMed: 26395554]
Publication Details
Author Information and Affiliations
Faculty of Medicine and University Hospital Cologne
Cologne, Germany
Federico II University of Naples
Naples, Italy
Neuromuscular Service
Evelina's Children Hospital
Guy's and St Thomas' Hospitals NHS Foundation Trust;
Randall Centre for Cell and Molecular Biophysics
Muscle Signalling Section
Faculty of Life Sciences and Medicine
King's College London
London, United Kingdom
Faculty of Medicine and University Hospital Cologne;
Max-Planck-Institute for Biology of Ageing;
Cologne Excellence Cluster on Cellular Stress Responses in Aging Associated Diseases (CECAD)
Cologne, Germany
Evelina's Children Hospital
Guy's and St Thomas' Hospitals NHS Foundation Trust;
Randall Centre for Cell and Molecular Biophysics
Faculty of Life Sciences and Medicine
King's College London
London, United Kingdom
Publication History
Initial Posting: March 21, 2024.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Möller B, Coppola A, Jungbluth H, et al. DYNC1H1-Related Disorders. 2024 Mar 21. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
%20and%20DYNC1H1-related%20neurodevelopmental%20disorder%20(DYNC1H1-NDD%3B%20indicated%20in%20yellow).&p=BOOKS&id=601997_dync1h1-dis-Image001.jpg "Click on image to zoom")